Myocardial infarctions, more commonly known as heart attacks, generally occur when a thrombus (a clot, or an aggregate of debris, red blood cells, and platelets trapped in a network of fibrin protein) creates an arterial obstruction due to atherosclerotic plaque. The resulting severe ischemia (low or absent blood flow) sets up a serious supply deficit for the very metabolically active heart tissues. Beginning about 20 minutes into ischemia, a wave of cellular death rolls through the now stagnant perfusion field, from the innermost cardiac layers toward the outermost. Over the ensuing three to six hours, cell death is profound, with cardiac muscle cells, vascular cells, resident fibroblasts, and nerve cells all affected. There is a prototypical sequence of events related to a myocardial infarction:
Anoxia – When the occlusion occurs, blood flow is reduced significantly. If the ischemia is profound enough, there will be a functional absence of oxygen availability, or anoxia. The shorter the period of anoxia, the less damage to the cardiac tissues. Within 20 minutes of anoxia, permanent damage has begun.
Necrosis – If the period of anoxia is extended beyond a cell’s reserve capacity, cell death results. Exhaustion of available aerobic and anaerobic metabolic substrates, increased toxicity from failed removal of cellular wastes, and more make cellular survival impossible. The duration of this stage begins at about 20 minutes post-blockage and can continue for about six hours, at which time all at-risk cells in the perfusion field will have succumbed.
Granulation – Neutrophils and then macrophages (two cell types that contribute to the body’s immune system) infiltrate the infarcted tissues at around four to 12 hours post-event. Neutrophils and macrophages engulf and remove the degrading dead cells. This prepares the region for subsequent remodeling that will replace cells and tissue architecture. At the same time, fibroblasts from within the infarcted zone and from unaffected proximal areas migrate to the necrotic zone and begin proliferating and secreting fibronectin, a type of loose connective tissue protein. The activities of these cell types — removing necrosis, adding fibronectin, and other activities that are precursors to scarring — remodels the infarcted tissue, which takes on a roughly granular appearance. Hence the name of this stage.
Scarring – The same cells active in granulation remain so for quite some time. For approximately two months after the infarction event, a robust scar tissue will develop, replacing the once viable and contractile cardiac tissues. The scar is largely a function of fibroblast cells altering their production, exuding less fibronectin and adding much more collagen into the extracellular spaces and matrices.
This sequence of processes profoundly affects the heart’s morphology beyond the localized cellular changes. The walls of the ventricles, the most common sites of infarction, are thickly muscled and replete with conductive cells driving the heartbeat. As we progress from time zero to the completion of scarring, dramatic changes occur in the shape, appearance, and function of the ventricles.
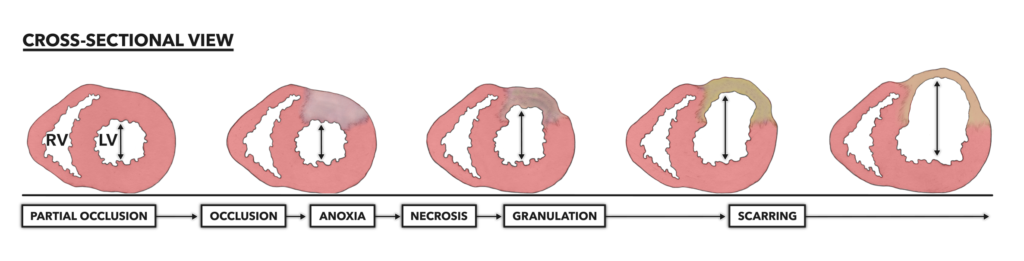
Figure 1 – Infarctions are always preceded by a pathology, most frequently a long-term and largely outwardly asymptomatic disease called atherosclerosis. Atherosclerosis slowly chokes the diameter of a coronary artery, making it susceptible to more severe and complete obstruction. Once occlusion occurs, myocardial infarction follows. Note that scarred tissue cannot contract. This means the affected ventricle will not be able to completely eject blood, even with remaining muscular contraction. Pink coloration = viable myocardium. RV = Right ventricle. LV = Left ventricle.
Myocardial infarction results in the thinning of the infarcted ventricle over time. As the wall thins, the chamber diameter inside increases. As remodeling and scarring occur, overall heart diameter also increases. This condition is known as pathological cardiac hypertrophy. The architectural changes increase mechanical stresses on the ventricular wall and a systematic decrease in contractile function. This means the ventricle is no longer able to contract with its full circumference; its contractions become asymmetrical. Cardiac output is significantly reduced due to scar-induced asymmetric contraction. The extent of functional loss after infarction is directly correlated to the amount of myocardium lost and scarred.
The sequence of events that follows a complete occlusion differs from that of an incomplete occlusion:
Complete occlusion → Anoxia → Necrosis → Granulation → Scarring & Impaired function
Incomplete occlusion → Hypoxia → Angiogenesis & Preserved function
Note that hypoxia — low but not absent oxygen availability — drives the formation of new capillaries and vessels within the margins of the affected perfusion field. The new vessels are driven to form from vessels higher in the perfusion field or from vessels in closely bordering perfusion fields. These additional small vessels supply blood to the hypoxic perfusion field, keeping some of the affected cardiac tissues viable and functioning normally. These new sets of vessels are called collateral vessels. They are similar to alternative routes around a traffic jam.
The effects of hypoxia on cardiac vessel angiogenesis is quite interesting. If we can create hypoxia in a normally functioning heart, we might be able to drive angiogenesis and expand the number of vessels delivering blood to all the heart’s tissues. This would create an infarction-resistant heart.
How could one induce and titrate the dose of cardiac hypoxia needed to drive angiogenesis? Exercise. Low doses of exercise have been demonstrated to enhance collateralization in both healthy and diseased hearts. One can conjecture that programmed and regular higher intensity exercise that pushes an athlete into more profound and transient systemic hypoxia may be effective in driving collateral growth and protecting the heart from future damage. While we can assemble a strong data-driven argument to support this conjecture, most research in this area is performed on ponies, pigs, and rats, who generally don’t or won’t do CrossFit in the laboratory.
Additional Reading
- The Heart, Part 1: Location & Orientation
- The Heart, Part 2: Muscular Organization
- The Heart, Part 3: Muscular Composition & Arrangement
- The Heart, Part 4: Conductive Pathway
- The Heart, Part 5: Blood Flow
- The Heart, Part 6: Blood Vessel Basics
- The Heart, Part 7: Coronary Circulation
- The Heart, Part 8: Systemic and Pulmonary Circulation
- The Heart, Part 9: Blood
- The Heart, Part 10: Myocardial Infarction
Comments on The Heart, Part 11: Myocardial Remodeling and Salvage
HOW I GOT MY HUSBAND BACK WITH THE HELP OF DR LARRY CONTACT ON WHATS_APP NUMBER +1(424)-261-8520
Greetings to you all. It was two years ago I lost a beautiful relationship with my husband and ever since then I have been trying all possible best to get him back but it hasn't worked out for me. But today it's a different story after I got a contact from the Internet about this wonderful love spell caster Dr Larry. I contacted him by email. I finally told him all my problems and he said to me, i will help you. I was happy because I have read testimonies about people he has helped without complications. Just in 48hrs my husband called me sounding so loving and romantic on the phone and told me he is coming home to me and he wants us to take our marriage vows again to prove how sorry he was.. Dr Larry helped me to bring my husband back to me and also helped me with fertility spells, so today I am a mother. Since last year I was shy to come out publicly but after giving birth a few weeks ago I chose to let the world know about a great man called Dr Larry. Contact him, With his powerful spell you shall find peace.
Email. assurancesolutionhome@gmail.com
whatsapp +1(424)-261-8520
CrossFit saved my wife's life - a "Widow Maker" heart attack survivor story.
My wife (Cynthia) and I started CrossFit in 2012 and we opened CrossFit Point A in 2015. At that time, Cynthia did trail running every morning with the dogs and helped me with CrossFit classes during the evening. On July 20th, 2015, Cynthia ran in the morning and worked out with me at lunch time. Later that evening, she worked out with her girlfriend at our box and sat down on the ground to change her shoes.
Cynthia fell backwards and I was close enough to catch her...I thought she fainted or had a seizure.
Her breathing slowed to a stop, the blood drained from her face and I began rescue breathing. Her pulse began to slow and fade away and it too eventually stopped. I began chest compressions. Being a police officer, my training kicked in. I directed our younger son to call 911and our older son to clear the parking lot and direct Fire Rescue in.
Cynthia was without any signs of life for over 20 minutes, however a heartbeat was restored as we pulled into the Emergency Room, where she was rushed into the Cath-Lab.
We had no idea that she had severe heart disease, a 100% blocked artery and two other arteries with 70% blockage each...very bad news. Cynthia was in a coma for 3 days and family was called to the hospital say goodbye. This was the toughest 3 days of my life, watching my wife breathing through a tube, fighting for her life...with a chest tube, pneumonia and collapsed lung.
After the third day, Cynthia's meds were reduced and she responded positively to testing. Her doctors asked Cynthia, if she could hear them, to move her foot. I stared at her foot and it move about a half an inch! It was a sign that she was coming back...
Since then, Cynthia has re-learned to talk, walk, drive on her own, go back to work and CrossFit again. It's been a tough and trying time with many ups and downs. Cynthia's doctors have all commented on how she is a living miracle.
I am married to the toughest person I know, with the biggest heart...
When she was brought into the Emergency Room in 2015, I could hear the staff as they whispered to each other, "That's the CrossFit girl" which I took as a negative...somehow that CrossFit was to blame for her condition. They were right. It was CrossFit that was to blame for her survival and her recovery.
I am proud to say that we are the owners of a CrossFit Affiliate and we believe that CrossFit saves lives.
I had a cardiac arrest (in the box) 18 months into Crossfit at age 54.
First responder box members and emergency room staff saved my life with CPR and emergency stenting but I remember my Cardiologist saying that getting fit through Crossfit meant I had the resilience to survive long enough for the catheter surgeons to do their work.
He also speculated that, by working my heart hard against the (then partial) blockage in my coronary artery, I had started to "grow my own bypass" in the year or so of Crossfit prior to my arrest. This keys in to the article above. Before discharging me, he said that, I had survived with an apparently undamaged heart muscle and should get back to Crossfit as it was keeping me alive.
I was back in the box with 3 months and will celebrate 8 years as a Crossfitter early in 2021. I now coach at my local box and am about to introduce some classes specifically for older adults.
Awesome story! Thanks for sharing.
Interesting theory. I wish we could take a peek inside the hearts of Games athletes.
The Heart, Part 11: Myocardial Remodeling and Salvage
7