Our lack of progress in developing effective treatments for the majority of cancers (outside of breast, colon, and lung cancers, and some liquid tumors) suggests that the dominant framework used to understand the origin and progression of cancer — and so its potential causes and treatments — is flawed. The rationale for precision medicine, gene-targeted therapies, and immunotherapies is based on the premise that cancer is a genetic disease according to the somatic mutation theory (SMT).
Unfortunately, this lack of progress in cancer treatment has been met not by a diversification of efforts but by a concentration on a limited scope of research based on the SMT, crowding out alternative hypotheses. Worse still, there is evidence to suggest that many of the treatments developed through this framework are harmful, as was recently highlighted in a review of the multiple failed therapeutic approaches used to manage glioblastoma (57). New immunotherapies can accelerate the growth rate of some cancers and actually expedite patient mortality (58). Although many people survive cancer by using conventional cancer therapies, many also pay a high price as new health issues arise out of the therapies they received. “Cancer survivor medicine” is emerging as a new medical field to address the multitude of health issues that afflict cancer survivors, including depression fatigue, pain, neuropathy, lymphedema, sleep issues, weight gain, cognitive dysfunction, sexual dysfunction, and fear of recurrence (59). Most cancer survivor health issues arise indirectly from therapies based on the gene theory of cancer.
Clearly, the stakes are high and demand a careful consideration of our current understanding of cancer and its origins. It is unlikely, if not impossible, that effective cancer therapies will be developed if the origin of the disease is misunderstood. To that end, over the past several weeks, we have contrasted the SMT with the mitochondrial metabolic theory of cancer (MMT).
The MMT Is the Most Compelling Explanation for the Origin of Cancer
The credibility of a theory’s explanation of a complicated phenomenon is dependent on the extent to which the theory can explain the facts associated with the phenomenon (56). The MMT can better explain critical hallmarks of cancer than can the SMT.
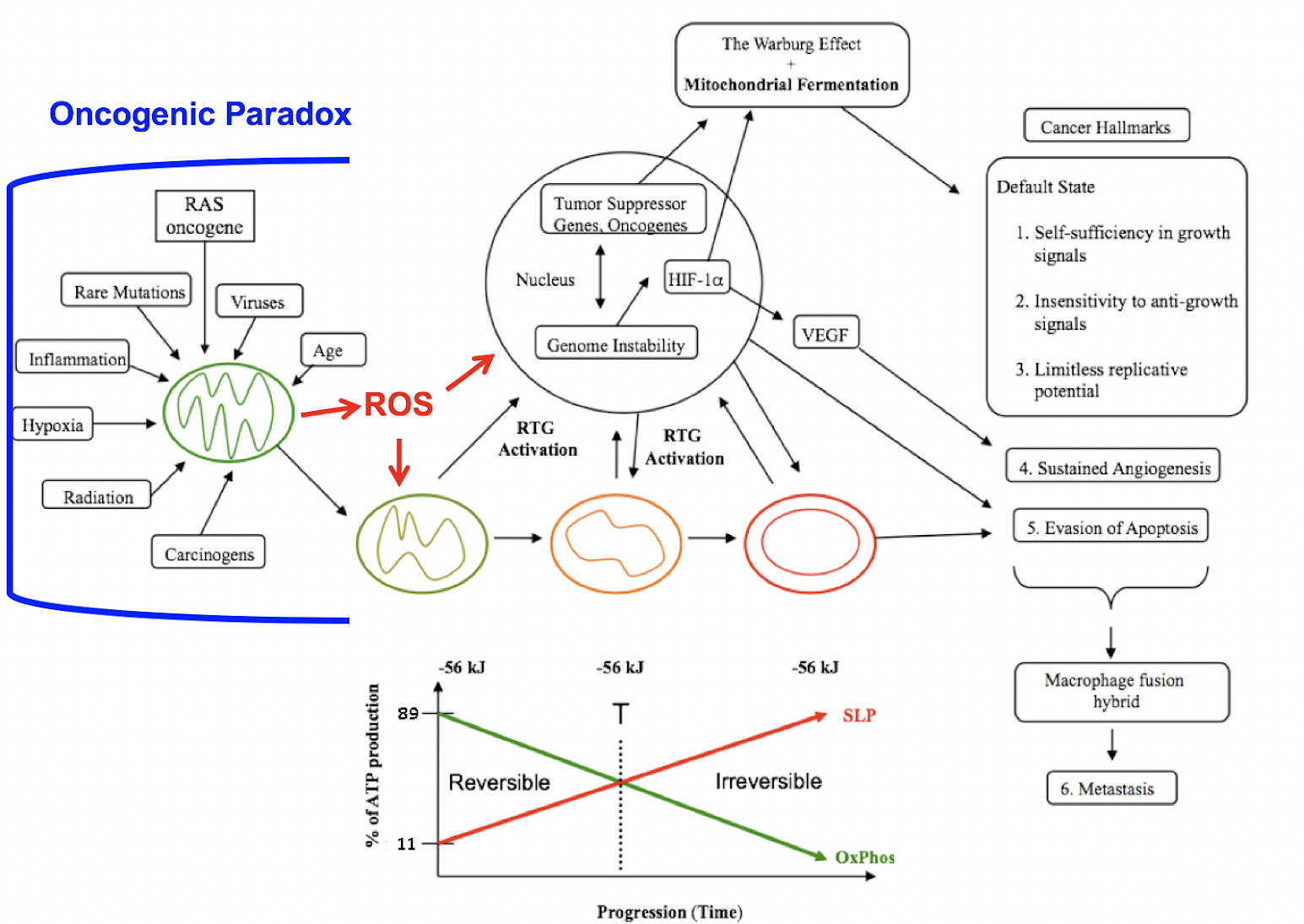
Figure 1: Mitochondrial respiratory dysfunction as the origin of cancer. Reprinted with modifications from (60).
Figure 1 shows how mitochondrial dysfunction as an initial event can account for the major hallmarks of cancer.
Cancer can arise from any number of non-specific events that damage the respiratory capacity of cells over time. These events can include carcinogens, radiation, intermittent hypoxia, chronic inflammation, rare inherited mutations, oncogenes, oncogenic viruses, and age. Albert Szent-Györgyi originally referred to the phenomenon by which a broad range of non-specific events could cause cancer through a common pathophysiological mechanism as the “oncogenic paradox” (61). Siddhartha Mukherjee also struggled to explain the oncogenic paradox in his Pulitzer Prize-winning book, The Emperor of All Maladies (62). Thomas Seyfried has shown that the common pathophysiological mechanism linking the non-specific events to cancer is oxidative respiratory dysfunction, thus solving the oncogenic paradox (56, 63).
The path to carcinogenesis will occur only in those cells capable of enhancing energy production through aerobic fermentation (substrate-level phosphorylation, or SLP). Although aerobic fermentation was thought to involve only glucose (the Warburg effect), Christos Chinopoulos and Seyfried recently showed that glutamine also could drive energy production in cancer cells through SLP at the succinyl CoA ligase step in the mitochondria (18). Despite the shift from respiration to SLP, the ΔG’ of ATP hydrolysis remains fairly constant at approximately -56 kJ, indicating that the energy from SLP compensates for the reduced energy from OxPhos (Figure 1). Indeed, Chinopoulos and Seyfried consider mitochondrial SLP to be the major source of ATP production in some cancer cells. The replacement of OxPhos with SLP ultimately leads to the formation of tumors. This is directly connected to abnormalities in the number, structure, and function of mitochondria (transition from green to red in Figure 1).
The mitochondrial stress response or retrograde signaling (RTG) will initiate the oncogene upregulation and tumor suppressor gene inactivation that are needed to maintain the viability of incipient cancer cells when respiration becomes incapable of maintaining energy homeostasis. The non-specific events that damage respiratory function produce reactive oxygen species (ROS) that are both mutagenic and carcinogenic (64,65). Genomic instability thus will arise as a secondary consequence of protracted mitochondrial stress from disturbances in the intracellular and extracellular microenvironment (66). Oncogenes are simply facilitators of tumor formation as they upregulate those pathways needed for fermentation metabolism.
In other words, the genomic defects seen in tumor cells arise as effects rather than causes of cancer.
Defects in the number, structure, and function of mitochondria can account for the six major hallmarks of cancer as outlined by Douglas Hanahan and Robert Weinberg (2). As normal mitochondrial function maintains the differentiated state of quiescence, a loss of OxPhos leads to the default state of proliferation. Carlos Sonnenschein and Ana Soto showed that proliferation rather than quiescence is the default state of all metazoan cells (67). A return to the default state of proliferation thus can explain the first three cancer hallmarks in Figure 1. As cancer cells ferment, they acidify the microenvironment, thus leading to sustained vascularization or angiogenesis (hallmark 4). This is consistent with the view that many solid tissue cancers behave as unhealed wounds (68). As mitochondria control the cellular kill switch (programmed cell death or apoptosis), defects in mitochondrial function can cause evasion of apoptosis (hallmark 5). Metastasis is the primary cause of mortality in most cancers.
According to the MMT, metastasis arises from respiratory damage in cells of myeloid/macrophage origin (41). See Figure 2 below (published with additional detail in Part 4 of this series on 190208).
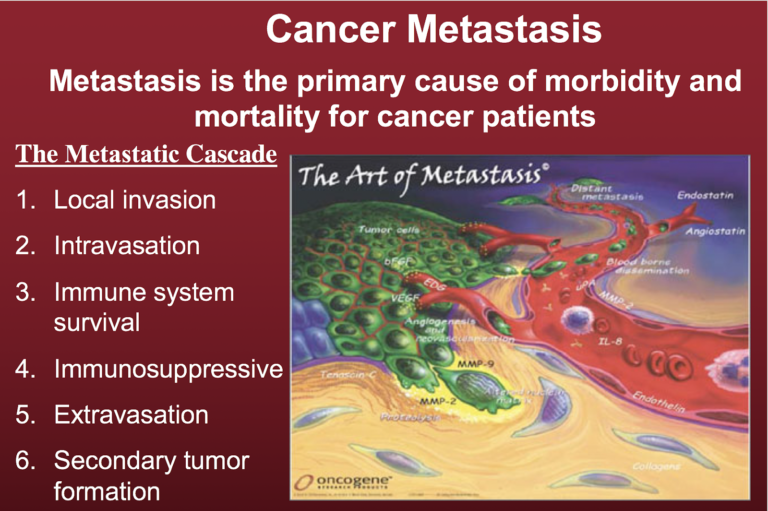
Figure 2. The metastatic cascade. A key aspect of metastasis is the non-random manner by which the metastatic cancer cells invade distant organs and tissues, with lung, liver, and bone as some of the more preferred sites. The green cells depicted here are the tumor cells.
The degree of malignancy is linked directly to the energy transition from OxPhos to substrate-level phosphorylation. This scenario links all major cancer hallmarks to an extrachromosomal respiratory dysfunction and can explain the origin of cancer better than can the SMT (56). The T in Figure 1 signifies an arbitrary threshold where the shift from OxPhos to SLP might become irreversible.
Therapeutic Implications for the Mitochondrial Metabolic Origin of Cancer
If cancer is a mitochondrial metabolic disease rather than a disease of somatic mutation, there are significant implications for successful treatment and therapies. Our interest in this series is to explore the evidence for or against the SMT and MMT rather than present a thorough investigation into potential therapies. We note, however, that additional research by Seyfried and Dominic D’Agostino, among others, indicates that metabolic therapies based on the MMT (and considering cancer as a systemic disease regardless of the specific tissue or organ system containing invasive or metastatic tumor cells) may target the metabolic abnormalities in tumor cells while enhancing the metabolic efficiency in normal cells (55, 69). This type of investigation into alternative therapies underscores the need for an accurate understanding of cancer’s nature and origins.
Conclusion
Once again, we note that the credibility of a theory’s explanation of a complicated phenomenon is dependent on the extent to which the theory can explain the facts associated with the phenomenon. The evidence presented in this series indicates that the mitochondrial metabolic theory can explain the facts of cancer better than the somatic mutation theory. An accurate understanding of the nature of cancer is essential to addressing the current cancer crisis via effective therapies.
Related
Is Cancer a Genetic or Metabolic Disease? Part 1
Is Cancer a Genetic or Metabolic Disease? Part 2
Is Cancer a Genetic or Metabolic Disease? Part 3
Is Cancer a Genetic or Metabolic Disease? Part 4
Is Cancer a Genetic or Metabolic Disease? Part 5

Thomas N. Seyfried is professor of biology at Boston College. He received a doctorate in genetics and biochemistry from the University of Illinois—Urbana-Champaign in 1976. He did his undergraduate work at the University of New England, where he recently received the distinguished Alumni Achievement Award. He also holds a master’s degree in genetics from Illinois State University. Seyfried served with distinction in the United States Army’s 1st Cavalry Division during the Vietnam War and received numerous medals and commendations.
He was a postdoctoral fellow in the Department of Neurology at the Yale University School of Medicine and then served on the faculty as an assistant professor in neurology. Seyfried previously served as chair of the Scientific Advisory Committee for the National Tay-Sachs and Allied Diseases Association. He recently received a Lifetime Achievement Award from the Academy of Complementary and Integrative Medicine and the Uncompromising Science Award from the American College of Nutrition for his work on cancer.
He presently serves on several editorial boards, including those for Nutrition & Metabolism, Neurochemical Research, the Journal of Lipid Research, and ASN Neuro. Seyfried has over 180 peer-reviewed publications and is author of the book “Cancer as a Metabolic Disease: On the Origin, Management, and Prevention of Cancer” (Wiley Press).
References
Note: These references include those previously published in “Is Cancer a Genetic or Metabolic Disease? Part 1,” “Part 2,” “Part 3,” “Part 4,” and “Part 5.”
- Siegel RL, Miller KD, and Jemal A. Cancer statistics, 2018. CA: A Cancer Journal for Clinicians 68.1(2018): 7-30. Available here.
- Hanahan D and Weinberg RA. Hallmarks of cancer: the next generation. Cell 144.5(2011): 646-674. Available here.
- Vogelstein B, Papadopoulos N, Velculescu VE et al. Cancer genome landscapes. Science 339.6127(2013): 1546-1558. Available here.
- Hou JP and Ma J. DawnRank: discovering personalized driver genes in cancer. Genome Medicine 6.7(2014): 56. Available here.
- Iranzo J, Martincorena I, and Koonin EV. Cancer-mutation network and the number and specificity of driver mutations. Proceedings of the National Academy of Sciences of the United States of America 115.26(2018): E6010-E6019. Available here.
- Fearon ER and Vogelstein B. A genetic model for colorectal tumorigenesis. Cell 61.5(1990): 759-767. Available here.
- Tomasetti C and Vogelstein B. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science 347.6217(2015): 78-81. Available here.
- Vaux DL. In defense of the somatic mutation theory of cancer. BioEssays 33.5(2011): 341-343. Available here.
- McLeod HL. Cancer pharmacogenomics: early promise, but concerted effort needed. Science 339.6127(2013): 1563-1566. Available here.
- Ju J, Zhu A, and Yuan P. Progress in targeted therapy for breast cancer. Chronic Diseases and Translational Medicine 4.3(2018): 164-175. Available here.
- Seyfried TN. Cancer as a Metabolic Disease: On the Origin, Management and Prevention of Cancer. Hoboken, New Jersey: John Wiley & Sons, Inc., 2012. Available here.
- John AP. Dysfunctional mitochondria, not oxygen insufficiency, cause cancer cells to produce inordinate amounts of lactic acid: the impact of this on the treatment of cancer. Medical Hypotheses 57.4(2001): 429-431. Available here.
- Kim A. Mitochondria in cancer energy metabolism: culprits or bystanders? Toxicological Research 31.4(2015): 323-330. Available here.
- Pelicano H, Zhang W, Liu J et al. Mitochondrial dysfunction in some triple-negative breast cancer cell lines: role of mTOR pathway and therapeutic potential. Breast Cancer Research 16.5(2014): 434. Available here.
- Srinivasan S, Guha M, Dong DW et al. Disruption of cytochrome c oxidase function induces the Warburg effect and metabolic reprogramming. Oncogene 35.12(2016): 1585-1595. Available here.
- Stefano GB and Kream RM. Cancer: mitochondrial origins. Medical Science Monitor 21(2015): 3736-3739. Available here.
- Warburg O. On the origin of cancer cells. Science 123.3191(1956): 309-314. Available here.
- Chinopoulos C and Seyfried TN. Mitochondrial substrate-level phosphorylation as energy source for glioblastoma: review and hypothesis. ASN Neuro 10(2018): 1-27. Available here.
- Boveri T. Concerning the origin of malignant tumours by Theodor Boveri. Translated and annotated by Henry Harris. Journal of Cell Science 121. Suppl.1(2008): 1-84. Available here.
- Nowell PC. The clonal evolution of tumor cell populations. Science 194.4260(1976): 23-28. Available here.
- Weinberg RA. The Biology of Cancer. New York City, New York: Garland Science, 2007. Available here.
- Darlington CD. The plasmagene theory of the origin of cancer. British Journal of Cancer 2.2(1948): 118-126. Available here.
- Rous, P. Surmise and fact on the nature of cancer. Nature 183(1959): 1357-1361. Available here.
- Baker SG and Kramer BS. Paradoxes in carcinogenesis: new opportunities for research directions. BMC Cancer 7(2007): 151. Available here.
- Soto AM and Sonnenschein C. The somatic mutation theory of cancer: growing problems with the paradigm? Bioessays 26.10(2004): 1097-1107. Available here.
- Sonnenschein C and Soto AM. Somatic mutation theory of carcinogenesis: why it should be dropped and replaced. Molecular Carcinogenesis 29.4(2000): 205-211. Available here.
- Burgio E and Migliore L. Towards a systemic paradigm in carcinogenesis: linking epigenetics and genetics. Molecular Biology Reports 42.4(2015): 777-790. Available here.
- Seyfried TN. Cancer as a mitochondrial metabolic disease. Frontiers in Cell and Developmental Biology 3(2015): 43. Available here.
- Li L, Connelly MC, Wetmore C et al. Mouse embryos cloned from brain tumors. Cancer Research 63.11(2003): 2733-2736. Available here.
- McKinnell RG, Deggins BA and Labat DD. Transplantation of pluripotential nuclei from triploid frog tumors. Science 165.3891(1969): 394-396. Available here.
- Hochedlinger K, Blelloch R, Brennan C et al. Reprogramming of a melanoma genome by nuclear transplantation. Genes & Development 18.15(2004): 1875-1885. Available here.
- McNutt M. Journals unite for reproducibility. Science 346.6210(2014): 679. Available here.
- Baker M. 1,500 scientists lift the lid on reproducibility. Nature 533.7604(2016): 452-454. Available here.
- Nosek BA and Errington TM. Making sense of replications. eLife 6: e23383, 2017. Available here.
- Sonnenschein C and Soto AM. Theories of carcinogenesis: an emerging perspective. Seminars in Cancer Biology 18.5(2008): 372-377. Available here.
- Fidler IJ. The pathogenesis of cancer metastasis: the ‘seed and soil’ hypothesis revisited. Nature Reviews Cancer 3.6(2003): 453-458. Available here.
- Tarin D. Cell and tissue interactions in carcinogenesis and metastasis and their clinical significance. Seminars in Cancer Biology 21.2(2011): 72-82. Available here.
- Garvin S, Oda H, Arnesson LG et al. Tumor cell expression of CD163 is associated to postoperative radiotherapy and poor prognosis in patients with breast cancer treated with breast-conserving surgery. Journal of Cancer Research and Clinical Oncology 144.7(2018): 1253-1263. Available here.
- Gast CE, Silk AD, Zarour L et al. Cell fusion potentiates tumor heterogeneity and reveals circulating hybrid cells that correlate with stage and survival. Science Advances 4.9(2018): eaat7828. Available here.
- Pawelek JM and Chakraborty AK. The cancer cell–leukocyte fusion theory of metastasis. Advances in Cancer Research 101(2008): 397-444. Available here.
- Seyfried TN and Huysentruyt LC. On the origin of cancer metastasis. Critical Reviews in Oncogenesis 18.1-2(2013): 43-73. Available here.
- Pawelek JM and Chakraborty AK. Fusion of tumour cells with bone marrow-derived cells: a unifying explanation for metastasis. Nature Reviews Cancer 8.5(2008): 377-386. Available here.
- Chernet BT, Fields C and Levin M. Long-range gap junctional signaling controls oncogene-mediated tumorigenesis in Xenopus laevis embryos. Frontiers in Physiology 5(2015): 519. Available here.
- Chernet BT and Levin M. Transmembrane voltage potential of somatic cells controls oncogene-mediated tumorigenesis at long-range. Oncotarget 5.10(2014): 3287-3306. Available here.
- Baker SG. A cancer theory kerfuffle can lead to new lines of research. JNCI Journal of the National Cancer Institute 107.2(2015): dju405. Available here.
- Kiebish MA and Seyfried TN. Absence of pathogenic mitochondrial DNA mutations in mouse brain tumors. BMC Cancer 5(2005): 102. Available here.
- Parsons DW, Jones S, Zhang X et al. An integrated genomic analysis of human glioblastoma multiforme. Science 321.5897(2008): 1807-1812. Available here.
- Martincorena I and Campbell PJ. Somatic mutation in cancer and normal cells. Science 349.6255(2015): 1483-1489. Available here.
- Martincorena I, Fowler JC, Wabik A et al. Somatic mutant clones colonize the human esophagus with age. Science 362.6417(2018): 911-917. Available here.
- Nishioka M, Bundo M, Iwamoto K et al. Somatic mutations in the human brain: implications for psychiatric research. Molecular Psychiatry 2018. Available here.
- Gauci ML, Boudou P, Baroudjian B et al. Occurrence of type 1 and type 2 diabetes in patients treated with immunotherapy (anti- PD-1 and/or anti-CTLA-4) for metastatic melanoma: a retrospective study. Cancer Immunology, Immunotherapy 67.8(2018): 1197-1208. Available here.
- Stupp R, Hegi ME, Mason WP et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. The Lancet Oncology 10.5(2009): 459-466. Available here.
- Johnson BE, Mazor T, Hong C et al. Mutational analysis reveals the origin and therapy-driven evolution of recurrent glioma. Science 343.6167(2014): 189-193. Available here.
- Seyfried TN, Flores R, Poff AM et al. Metabolic therapy: a new paradigm for managing malignant brain cancer. Cancer Letters 356. 2 Pt. A(2015): 289-300. Available here.
- Seyfried TN, Yu G, Maroon JC and D’Agostino DP. Press-pulse: a novel therapeutic strategy for the metabolic management of cancer. Nutrition and Metabolism 14.19(2017). Available here.
- Seyfried TN. Mitochondrial respiratory dysfunction and the extrachromosomal origin of cancer. Cancer as a Metabolic Disease: On the Origin, Management, and Prevention of Cancer. Hoboken, NJ: John Wiley & Sons, 253-259, 2012. Available here.
- Polivka J Jr., Polivka J, Holubec L, Kubikova T, Priban V, Hes O, Pivovarcikova K, and Treskova I. Advances in Experimental Targeted Therapy and Immunotherapy for Patients with Glioblastoma Multiforme. Anticancer Res 37(2017): 21-33. Available here.
- Ferrara R, Mezquita L, Texier M, Lahmar J, Audigier-Valette C, Tessonnier L, Mazieres J, Zalcman G, Brosseau S, Le Moulec S, Leroy L, Duchemann B, Lefebvre C, Veillon R, Westeel V, Koscielny S, Champiat S, Ferte C, Planchard D, Remon J, Boucher ME, Gazzah A, Adam J, Bria E, Tortora G, Soria JC, Besse B, and Caramella C. Hyperprogressive Disease in Patients With Advanced Non-Small Cell Lung Cancer Treated With PD-1/PD-L1 Inhibitors or With Single-Agent Chemotherapy. JAMA Oncol 4(2018): 1543-1552. Available here.
- Viscuse PV, Price K, Millstine D, Bhagra A, Bauer B, and Ruddy KJ. Integrative medicine in cancer survivors. Curr Opin Oncol 29(2017): 235-242. Available here.
- Seyfried TN, and Shelton LM. Cancer as a metabolic disease. Nutr Metab (Lond) 7(2010): 7. Available here.
- Szent-Györgyi A. The living state and cancer. Proceedings of the National Academy of Sciences of the United States of America 74(1977): 2844-2847. Available here.
- Mukherjee S. The Emperor of All Maladies: A Biography of Cancer. New York: Scribner, 2010. pp. 285, 303, 333, 342.
- Seyfried, TN, Flores RE, Poff AM, and D’Agostino DP. Cancer as a metabolic disease: implications for novel therapeutics. Carcinogenesis 35(2014): 515-527. Available here.
- Chen Y, Zhang H, Zhou HJ, Ji W, and Min W. Mitochondrial Redox Signaling and Tumor Progression. Cancers (Basel) 8 (2016). Available here.
- Ma Y, Bai RK, Trieu R, and Wong LJ. Mitochondrial dysfunction in human breast cancer cells and their transmitochondrial cybrids. Biochimica et biophysica acta 1797(2010): 29-37. Available here.
- Desler C, Lykke A, and Rasmussen LJ. The effect of mitochondrial dysfunction on cytosolic nucleotide metabolism. J Nucleic Acids (2010). Available here.
- Sonnenschein C, and Soto AM. The Society of Cells: Cancer and the Control of Cell Proliferation. New York: Springer-Verlag,1999.
- Dvorak, HF. Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N. Engl. J. Med 315(1986): 1650-1659. Available here.
- Arens, NC, and West ID. Press-pulse: a general theory of mass extinction? Paleobiology 34(2008): 456-471. Available here.
All links accessed Feb. 20, 2019.