Hepatic de novo lipogenesis refers to the liver’s synthesis of new fat from other substances, often carbohydrate and specifically fructose. This review summarizes the role of hepatic de novo lipogenesis (DNL)—and thus the role of fructose—in the development and progression of fatty liver disease.
Nonalcoholic fatty liver disease, the most common cause of elevated liver enzymes and among the most common indications for liver transplantation, is characterized by a liver fat concentration of more than 5 percent (by weight). Hepatic fat accumulates in the liver from three sources: (i) dietary lipids, (ii) esterification of plasma free fatty acids (FFAs), and (iii) the production of dietary fat by the liver, or hepatic de novo lipogenesis. In hepatic DNL, dietary carbohydrates are broken down to pyruvate, converted to citrate, and used to synthesize long-chain fatty acids.
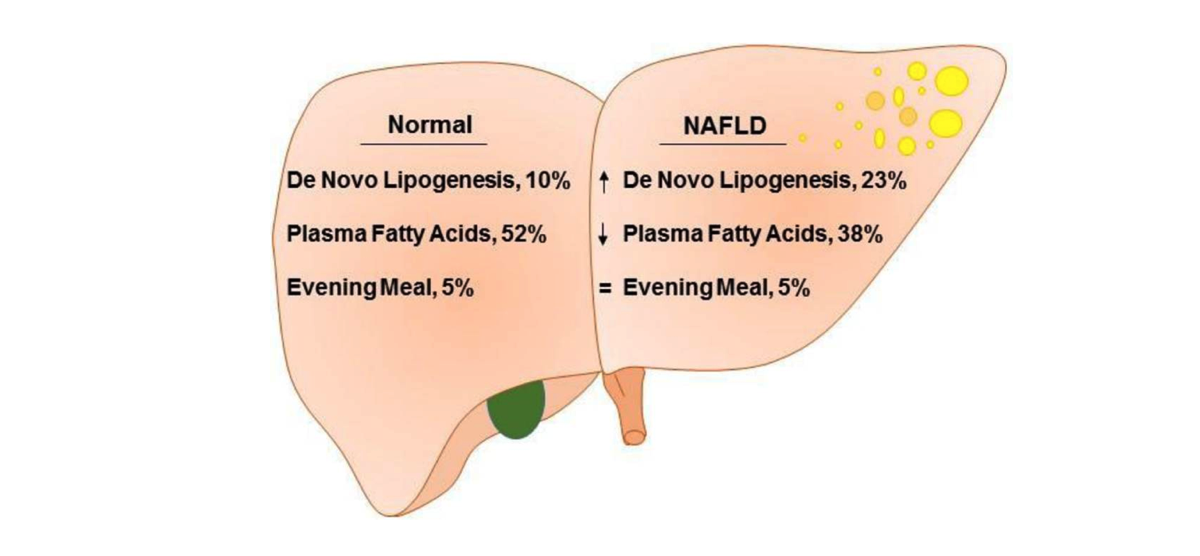
Figure 2 from Role of Dietary Fructose and Hepatic de novo Lipogenesis in Fatty Liver Disease. Lambert et al., used isotope analysis to compare de novo lipogenesis and fatty acid flux in obese subjects with and without NAFLD. The proportion of hepatic triglycerides deriving from the evening meal was not different, 5% versus 5%, at the end of the study. The proportion of triglycerides synthesized from plasma FFA in low liver-fat group (52%), was significantly higher than in high liver-fat group (38%). By contrast, triglycerides synthesized via de novo lipogenesis was significantly higher in high liver-fat group (23%), compared to low liver-fat group (10%).
Early studies suggested hepatic DNL was a minor contributor to liver fat stores, but these studies were done in fasting subjects; more recent research has shown that in obese patients with NAFLD, 26 percent of liver fat is derived from DNL (60 percent is from free fatty acids, 15 percent is from dietary fat). The share of liver triglycerides derived from hepatic DNL is higher in subjects with high liver fat, which suggests enhanced hepatic DNL is the primary defect in NAFLD. This has been supported by studies showing diets higher in carbohydrate promote DNL to a greater extent than diets lower in carbohydrate. Carbohydrate overfeeding, in particular, can lead to a rapid increase in liver fat.
Fructose uniquely promotes DNL, and thus uniquely promotes fatty liver disease. A variety of studies in both humans and animals have shown that increased fructose consumption is associated with greater risk of NAFLD and fructose restriction leads to NAFLD improvement. Fructose, once absorbed through the intestine, goes straight to the liver (unlike other macronutrients), where it upregulates multiple steps of the DNL process. While insulin promotes hepatic DNL in most cases, fructose (unlike glucose) does not require insulin to be metabolized. Finally, fructose may further increase metabolism through a variety of indirect mechanisms, including uric acid promotion and reactive oxygen species accumulation.
In sum, fatty liver disease is primarily a defect of excessive hepatic de novo lipogenesis, and fructose specifically promotes DNL through multiple mechanisms. Given these and other factors that suggest fructose plays a role in NAFLD progression, targeting fructose metabolism or intake may support NAFLD management.