
In the previous column (2), I reviewed some of the original studies of physiologist Ancel Keys (3, 4), who used weak associational data from population studies to develop twin hypotheses about how heart disease develops: the diet-heart and lipid hypotheses, originally proposed by John Gofman, MD. Whenever possible, Keys promoted those hypotheses as if they were solely his idea and both had been proven.
A key moment for Keys occurred at the 1955 Seminar of the World Health Organization’s (WHO) Study Group on Atherosclerosis and Ischaemic Heart Disease. There, Keys’ speculative conclusion that differences in blood cholesterol concentrations explained different rates of coronary heart disease in the six different countries he had studied (3) was vigorously contested, especially by Jacob Yerushalmy and Herman Hilleboe (5).
Unused to having his ideas challenged so forcefully, Keys left the meeting in a huff, determined to show the world and his critics he was correct.
He immediately set about raising the funds for a much larger study that would become known as the Seven Countries Study (SCS). Importantly, this was also an observational (non-randomized, non-interventional) study that could no more identify causative factors for coronary heart disease (CHD) than did the information he had already presented at the WHO seminar.
Perhaps Keys assumed that if he continued collecting and publishing enough circumstantial evidence with sufficient frequency, he and his like-minded acolytes who were even then gaining monopolistic control of the direction and funding of U.S. coronary heart disease (CHD) and atherosclerosis research would simply force the hypotheses to be accepted as fact.
History would prove Keys’ assumptions were not unreasonable. Today, his twin hypotheses are alive and well. In fact, they are thriving in medical schools around the globe. They continue to drive the direction of medicine in ways that perhaps even Keys could never have imagined.
Proving he had internalized at least some of the criticisms from Yerushalmy and Hilleboe’s lambasting, while Keys was setting up the SCS, he was also planning a remarkably well-designed intervention trial that would come to be known as the Minnesota Coronary Experiment (MCE).
This was a full-on randomized controlled trial (RCT) run under perfect experimental conditions. If ever Keys was going to prove his hypotheses as fact with the smoking gun evidence he required, the MCE was the experiment that would provide it.
Perhaps it might even earn him a Nobel Prize in Medicine?
Let us begin with a review of his epic study on the seven countries.
The Seven Countries Study (SCS)
In 1956, the U.S. Public Health Service awarded Keys with a grant for $200,000 per annum (6, p. 37). The funding would allow Keys to initiate a study of 12,770 middle-aged men in seven countries: Italy, Greece, Yugoslavia, Finland, the Netherlands, Japan, and the U.S. (7). Keys’ study was the first multi-country observational epidemiological study yet undertaken. But however large and expensive the study, it was, in the end, still just another observational (associational) study.
Observational studies cannot prove causation except in extremely unusual circumstances — e.g., when there is a single, very powerful causative agent such as chimney soot (8), cigarette consumption (9), or the cholera bacterium (10), as demonstrated in Figure 1.
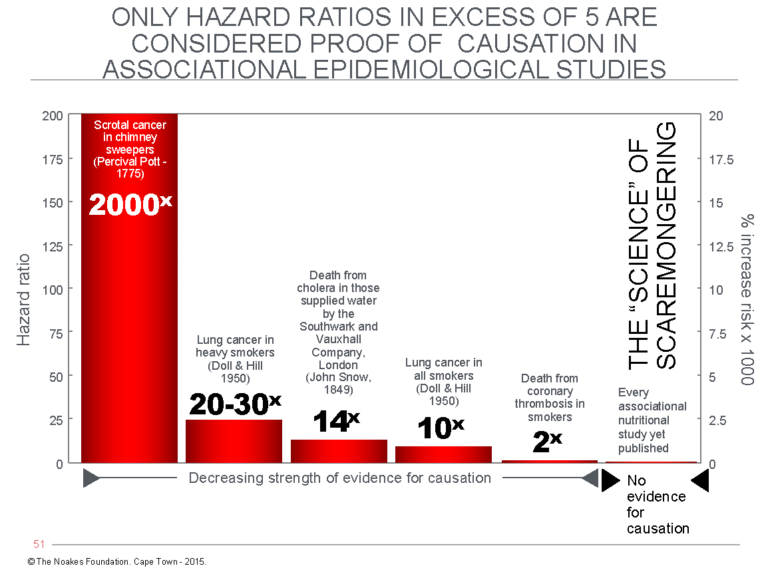
Figure 1: The hazard ratio can be used as a measure of the probability that evidence from an associational (observational) study is likely to be the result of a causal relationship. The hazard ratio compares the prevalence of persons experiencing a particular outcome when exposed to specific factors to the prevalence of the same conditions in a control group not exposed to each of those factors — for example, chimney soot in chimney sweepers (causing scrotal cancer) (8); cigarettes (causing lung cancer) (9); water contaminated with the Vibrio cholerae bacterium causing cholera (10); cigarettes (associated with heart attacks) (11); or different nutritionals (weakly and non-causally associated with a variety of conditions) (12). A hazard ratio in excess of 5 is generally accepted as proof of causation even in an associational study. Almost all nutritional associational studies produce hazard ratios of less than 1.4 and therefore cannot be considered proof of causation (12).
The SCS encountered numerous problems, succinctly outlined by Nina Teicholz (6, p. 36-42). Probably the worst was how the seven countries were selected for study. Instead of including countries that would challenge his hypothesis and might therefore disprove it (2, 13), Keys excluded countries such as France and Switzerland, which have high rates of fat intake but low rates of heart disease (2). Katherine Pett and colleagues (14) have recently argued, with good logic, that there were valid practical reasons why these countries could not be included. At least some of those explanations appear entirely credible. However, as P. D. P. Wood has shown (13), the selection of those particular countries introduced a bias into the study that can never be eliminated, even if those selections were initially made in good faith and not with the clandestine intent of biasing the results.
Keys once more included Japan, as the Japanese experience was crucial to his original publication (2, 3). The data for Japan reflected low rates of CHD and low dietary fat intakes, which anchored the bottom left-hand side of Keys’ iconic graph showing rising CHD rates with increasing dietary fat consumption (Figure 4 in reference 2). Without the Japanese data, the figure would not have looked as compelling.
But in the years after his completion of the SCS, Japan provided evidence that ran counter to Keys’ hypotheses: specifically, falling rates of heart disease despite increasing blood cholesterol concentrations (15) associated with an increase in fat intake from 13% to 33% of total calories in Japanese children (16).
Switzerland, in particular, would have been a most interesting country to include. Just as Keys’ hypotheses were taking off, Switzerland was providing evidence that its heart disease death rates were plummeting (Figure 2 – top panels), especially in women, just as the Swiss were starting to include more fat in their diets (Figure 2 bottom panel) (17). Swiss life expectancy also increased by five years among men and seven years among women during this period.
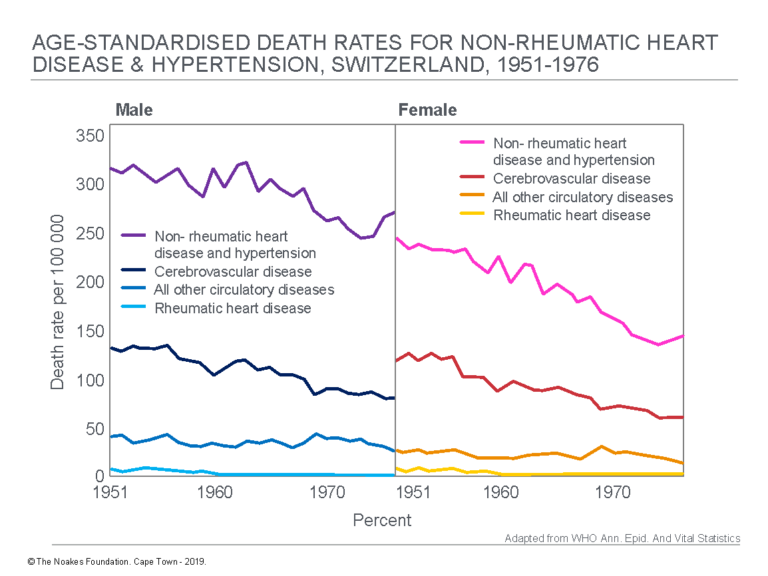
Figure 2: In the SCS, Keys did not include some countries known to have higher rates of fat consumption but low rates of CHD. The practical explanation for why this happened has been presented (14). Switzerland was one such country. A paper published in 1979 (17) showed death rates for CHD and stroke began to decline quite dramatically in men (top panel, left) and women (top panel, right) in Switzerland just as its citizens’ intakes of animal fats, pork, cheese, butter, and cream were increasing (bottom panel). Only the intake of milk showed a definite decline during this period. All this was happening at the same time the SCS was gathering momentum. Reproduced from reference 17, pp. 115, 118.
Figure 2 shows that just as Keys was starting to collect his data for the SCS, death rates for CHD and stroke began to decline in men and women in Switzerland (top panels). This decline was associated with increasing or stable intakes of animal fats, pork, vegetable fats, eggs, cheese, butter, and cream (bottom panel). The data were incompatible with Keys’ diet-heart hypothesis. Unfortunately, Switzerland was not included in any of Keys’ studies.
An equally large problem was, again (2), the manner in which the diets were evaluated. In the end, the diets of just 499 (4%) of the 12,770 SCS trial participants were studied. Keys attempted to hide this terminal weakness of the study, but this could not be hidden from investigative journalist Teicholz, who exposed it in her landmark book (6, p. 41). Teicholz explains, “If you read Keys’s study design very closely, you find that, of the 12,770 participants, the food they ate was evaluated for only 499 of them, or 3.9 percent. And there was no consistency among nations as to how the nutritional data were collected” (p. 40).
Almost as bad, Teicholz discovered that some of the data for the Greek participants was collected during Lent, when Greek Orthodox church tradition requires abstention from all foods of animal origin. As a result, the collected dietary data would have underestimated the true saturated fat intake in a population with a low rate of coronary heart disease. This would falsely favor Keys’ hypothesis, although Pett et al. (14) have argued that since followers of this religious order may restrict their food intake on as many as 200 days a year, Keys was justified in studying them on days they were restricting food intake in this way.
Pett et al. do not address the criticism that the diets of only about 4% of subjects were assessed in the SCS. This is important because these authors believe plant-based diets are the most healthy option for humans. Thus, they are keen to preserve the apparent integrity of what they believe was the primary finding of the SCS — that “saturated fat was correlated with heart disease” (14, p. 4). But if the dietary analyses in the SCS were poorly conducted and not representative of the total population under investigation, that conclusion is moot.
Then there was the problem that the contribution of “sweets,” meaning sugar products and pastries, to CHD risk was not analyzed until many years later (6, p. 42). Moreover, dietary data did not accurately measure sugar consumption and may have underestimated the contribution of differences in sugar intake to differences in CHD rates in the different countries. The analysis listed some carbohydrate-laden foods as if they contained predominantly saturated fats. Conversely some animal foods were listed as saturated fats when, in reality, they contained more unsaturated than saturated fats (38).
Zoë Harcombe et al. note:
The study classified cake and ice cream as saturated fats, as opposed to refined carbohydrates, an error which is repeated by contemporary food scientists. Meat and eggs are described as saturated fat when their fat content is primarily unsaturated. Butter and cream are one third unsaturated fat, which was not noted in their analysis. So here we have a profoundly influential research project introducing imprecise evaluations of macronutrients which have continued to the present day. (38, p. 241, my emphasis)
They continue:
There is a need to accurately define the macro and micronutrient content of food. Biscuits, savoury snacks and processed foods should not be defined as saturated fat because they are substantially carbohydrates. Red meat is not a saturated fat but a combination of various fatty acids. Sirloin steak for example is approximately 71% water, 21% protein, 3% unsaturated fat and 2% saturated fat. Natural foods such as meat, fish, eggs and nuts contain saturated, monounsaturated and polyunsaturated fats, only the proportions vary. Few people appreciate that it is impossible to eat saturated or polyunsaturated fat alone. Dairy products are the only food group with more saturated than unsaturated fat. Many of the foods demonized by past research groups, even lard, contain more unsaturated than saturated fat. Dietary fat consumption is a key provider of essential fats and fat soluble vitamins. (p. 241)
Harcombe et al. provide a simple example of the problems caused by the modern interpretation of the SCS findings: “Using a 100g steak, as an example, with 5.4g of fat, it is difficult to accept that the 39% of the fat which is saturated is damaging to the cardiovascular system while the 61% of the fat which is unsaturated is protective. Keeping in mind that the total fat content of the steak will provide all but 3 of the 13 vitamins and 16 minerals that are a prerequisite for the maintenance of good health” (p. 242).
An additional problem with the original study: Many of the countries included were still recovering from the deprivations of World War 2. These effects would likely have some impact on the outcomes Keys was measuring.
Keys ultimately had to acknowledge that the SCS dietary data were incomplete. He chose to publish the content in an obscure Dutch journal that had “very little circulation outside the Netherlands and even there (was) primarily read by nutritionists” (6, p. 41). This was not the only time Keys and his comrades would hide inconvenient data by either publishing the results in an obscure medical journal or simply not publishing the data at all.
Despite being highly flawed, the data Keys and his colleagues collected have proven influential and enduring. During the trial, the electrocardiograms, blood pressures, body masses, blood cholesterol concentrations, and diets of the participants were measured, and the individuals were followed for many years to determine if and when they developed complications of coronary artery disease or other causes of mortality. These data have continued to be analyzed and described ever since (18-25).
Major findings of the Seven Countries Study
The major findings of the SCS were reviewed in 1993 by two of the study’s principal investigators, Henry Blackburn, MD, and Alessandro Menotti, MD and Ph.D., in a special publication prepared to honor Keys’ 90th birthday (25).
But before we delve into those findings, it is critical to stress the authors’ disclaimer, added in the final two paragraphs of the article: “Obviously, an attempt to relate the 15-year risk of death of the individual to his diet is impracticable … . Those estimates of average contributions of the nutrients of interest should be reasonably good for the cohorts but nothing can be said about the risks of individual men … . The relationships shown here are statistically important but they are not claimed to be necessarily causal” (21, p. 914, my emphasis added).
In other words, the findings of the SCS cannot be used to justify nutrition guidelines for individuals, which means they also cannot be used to justify national dietary guidelines.
This disclaimer is, unfortunately, never mentioned by those who use Keys and his colleagues’ work to justify the prescription of national dietary guidelines.
Here, I reproduce in an edited format the conclusions the SCS authors considered the most important. Some of the results are for data generated from the cross-sectional population/country comparisons; the others make predictions based on the (non-randomized) prospective study that followed the health of the 12,770 participants over the first two and a half decades of the study.
- Age was the strongest risk predictor for coronary heart disease (CHD) incidence and all-cause mortality (p. 160-161).
- Diastolic blood pressure >95mmHg showed a strong relationship with CHD risk (p. 160).
- Men grouped within the top 20% of the age-specific blood pressures at entry had twice the incidence of CHD (p. 161).
- The strongest consistent relationship with CHD prevalence in the different populations/countries was the prevalence of initial serum cholesterol values greater than 250 mg/dL (6.5 mmol/L) in those populations (p. 160).
- The correlation of serum cholesterol concentrations with 10-year CHD death rates in the different populations/countries were highly significant (p. 161).
- All-cause death rates were only slightly correlated with average serum cholesterol concentrations in the different populations/countries (p. 161).
- Serum cholesterol concentrations were negatively (inversely) related to non-cardiovascular disease risk in different populations/countries (p. 161).
- Serum cholesterol concentrations greater than 220 mg/dL (5.7 mmol/L) were a “particularly important individual risk factor for CHD” (p. 161).
- Decreasing serum cholesterol concentrations in individuals “tended to be associated” with increasing rates of non-coronary deaths (p. 161).
- Electrocardiographic abnormalities, in particular the presence of abnormal Q waves (indicating, most commonly, a previous myocardial infarction, or heart attack), had “strong prognostic power” for the development of CHD (p. 161).
- In Italy, Greece, and Japan, cigarette smoking was a minor risk factor for deaths from all causes and CHD (p. 161).
- In all other countries, there was a strong individual relationship between smoking and both CHD and non-CHD causes of death (p. 161).
- In those countries, the risk of cancer rose linearly with the dose of cigarettes smoked (p. 161).
- Differences between 10-year CHD rates in the different populations/countries were not significantly related to average relative weight or rates of obesity (p. 161).
- Ten-year incidence of CHD in individuals was not significantly related to body mass.
- Ten-year death rates in individuals was linearly related to initial pulse rate.
- Risk factors had a different “force” in different populations and among individuals in those populations. Thus, the slope of the individual risk factor-CHD relationship was approximately twice as steep in the United States as in Europe, and in northern compared to southern Europe.
- Cigarette smoking had the greatest adverse effect on longevity. Elevated blood pressure had a lesser effect, whereas serum cholesterol concentration and body mass made “very little contribution.”
- Individual serum cholesterol concentrations were unrelated to the composition of the diet (p. 162).
Of note: This last statement, point 19, is written in a paragraph that is essentially unintelligible. It is written in the negative; in other words, the paragraph states the expected relationship between diet and serum cholesterol concentrations was not found. The authors attempt to spin this finding by providing a post-hoc explanation. Their explanation was that the diet varied too much among those (few meaning <4%) participants whose diets were actually recorded. Thus, in the opinion of the disappointed authors, to detect this variation would require multiple dietary surveys.
But how could the researchers know this if multiple dietary surveys — necessary to show this statement to be true — were never performed?
Post-hoc explanations of this kind are unacceptable. The study was testing the hypothesis that dietary fat intake is the principal determinant of the blood cholesterol concentration. When the study failed to find that, it effectively disproved the hypothesis that was being tested. Thus the sole conclusion honest scientists are allowed to draw from such an experiment is that their hypothesis was disproved. The next step required by the scientific process is for the researchers to develop a new hypothesis for further testing. That is how proper science is meant to work (26).
But Keys and his colleagues saw their responsibility differently. Keys was an activist and not a scientist. His goal was to wage war against cholesterol (27, p. 132). He fell in love with his hypothesis (and what it did for his global image (28)) and could simply not entertain the thought that he might be wrong. So if there were data that refuted his hypothesis, well then, it was obvious: The data were wrong and the persons providing the data were inferior scientists (29, 30).
Subsequently, A. Menotti and P. E. Puddu (31) confirmed: “The SCS never found a strong association of total fat intake with CHD and never promoted the idea of substituting total fat with carbohydrate” (31, p. 315, my emphasis). Instead, the authors claim the SCS was the first to test the dietary monosaturated/saturated fat ratio and show “its strong inverse association at ecologic level with CHD and all-cause mortality” (p. 315). The monosaturated/saturated fat ratio represented a kind of proxy, if elevated, of the Mediterranean diet’s use of dietary fat (p. 315).
In passing, Menotti and Puddu also stressed, “All the reports published by the SCS were always extremely cautious in attributing etiologic validity to ecologic associations.” In other words, the authors always understood association does not prove causation, so whatever Keys and his colleagues found could not be used to develop dietary guidelines for individuals. Not everyone yet understands this (14).
How did the SCS advance nutrition knowledge, if indeed it did?
The most important finding of the SCS was the incontestable confirmation that rates of coronary heart disease differed substantially in different countries. This suggests that environmental factors and not simply the aging process likely contribute to these differences.
The studies found that countries with higher rates of especially saturated fat intake (and correspondingly lower intakes of monosaturated fats) did indeed have higher CHD rates (20, 21) as Keys’ hypothesis predicted and as his defenders have correctly argued (14). But within individual countries were anomalies that were incompatible with Keys’ diet-heart hypothesis (6, p. 39). In addition, there is the abiding question of the accuracy of the dietary data collection and analysis.
Similarly, whereas CHD death rates were, as Keys’ lipid hypothesis predicts, indeed higher in countries and individuals with higher blood cholesterol concentrations, higher blood cholesterol concentrations were not associated with higher rates of all-cause mortality. This means deaths from other causes must have been reduced by high blood cholesterol concentrations. Additionally, all-cause mortality was not reduced in those with lower blood cholesterol concentrations. Instead, lower blood cholesterol concentrations were associated with increased rates of death from non-cardiac causes.
Thus, the study found that having a lower blood cholesterol concentration might well reduce the risk of dying from CHD, but it also increased the risk of dying from some other cause. Keys’ defenders (14) have conveniently ignored this inconvenient finding.
Additionally, longevity was not impaired by higher blood cholesterol concentrations. In fact, the populations with the greatest longevity were in the U.S. and Greece. Their longevity was unrelated to the amount of saturated fat or cholesterol they ate, or their blood cholesterol concentrations (6, p. 40). This finding is not mentioned in Blackburn and Menotti’s review of the major findings of the SCS (25).
And then, most inconveniently, dietary fat intake was unrelated to blood cholesterol concentrations. This is incompatible with the diet-heart and lipid hypotheses. Recall that Keys had based his original hypotheses on epidemiological, population-based studies. Now, using a more sophisticated study, he found evidence that disputed a number of his earlier conclusions.
According to the course of normal science, Keys should have acknowledged the findings of the SCS did not support his hypothesis that blood cholesterol concentration is determined by the amount of fat in the diet, or that low blood cholesterol concentration is the best surrogate marker, not just of heart health, but of total body, all-over health and longevity.
But by the time the SCS was published, such a retraction was no longer possible. Too much money and too many egos had been invested for anyone to acknowledge they had all been wrong (28). Instead, the messaging to the public continued to hide the truth — as it continues to do today.
How should these findings have been communicated?
One of Keys’ more devoted acolytes, Dr. F. H. Epstein, communicated the following message to the general public:
Drawing on all sources of data, the study has provided the strongest existing evidence that the risk of coronary heart disease is linked to the consumption of saturated fat, and that this relationship is mediated by serum cholesterol. This, of course, is the basis for the current conviction that reduction of saturated fat intake will lead to a reduction of coronary heart disease risk. (32, p. 170, my emphasis)
This interpretation is at best a half-truth that ignores clear evidence that (i) when studied in free-living humans, the diet did not influence the blood cholesterol concentration, and (ii) a low blood cholesterol concentration provided no overall, long-term health benefits.
The likely reason Epstein was asked to write this review was because Keys knew Epstein was too emotionally invested in the hypothesis to see or admit its flaws. If the diet-heart hypothesis had become a religion, Epstein was one of its most fervent disciples.
Epstein had already made up his mind 16 years earlier. In 1977, he wrote, “Though indirect, [the evidence supporting the diet-heart hypothesis] is so strong that the question of whether lowering serum cholesterol reduces the risk of coronary heart disease hinges no longer on the results of preventive trials alone.” He continued: “Preventive action in the community is justified now, without waiting for the results of preventive trials, however important they will be eventually to help the development of optimally effective programs” (33, p. 515). (One wonders whether, if he were alive today, he might want to modify that statement somewhat.)
Epstein used unusual logic to justify acting in the absence of evidence. First, he argued the negative results provided by the Coronary Drug Project (34), in which lowering cholesterol with two separate drugs failed to produce any beneficial outcomes, “in no way, shakes the lipid theory of atheroma formation (i.e., the lipid hypothesis)” (33, p. 518). “Consequently, whatever the method of intervention, an overall negative result for a trial as a whole does not rule out the possibility that intervention might be effective in such a sub-group,” he added (p. 518-519).
This effectively means there is no need for any more trials, since no trial can rule out the possibility that some sub-group of patients might benefit. So what Epstein really implied is that, even in the face of compelling negative evidence, we can justify continuing to give that tested intervention to everyone.
Epstein wrote:
There is every justification, therefore, to aim a major preventive effort on this fifth of the population at the highest risk in terms of serum cholesterol, blood pressure and smoking and to take multiple risks into account in designing preventive trials. Concentrating on those at highest risk will most likely facilitate adherence to the preventive program and, by ensuring a sizeable expected response to the prophylactic treatment, will enhance the probability of detecting a positive result if the method of intervention is indeed effective. (p. 519, my emphasis)
But hadn’t millions of dollars just been spent proving the intervention was not effective? What more evidence does one need that the theory is wrong?
Even more remarkably, he added: “Even a negative trial result, for reasons which were discussed, would in no way indicate that prevention by the methods now available, started sufficiently early in life, is ineffective, thus providing a further reason for not waiting” (p. 521).
Epstein’s arguments, no longer based on science, instead cross over into the realm of religious certainty. He claims the diet-heart and lipid hypotheses are facts, and no amount of negative evidence will dislodge that conviction. Perhaps Epstein did not understand the SCS was an associational study, which even Keys had not “claimed to be necessarily causal” (21, p. 914). But clearly, somewhere along the way, that subtlety was lost.
Epstein should have told the general public the following:
- The blood (serum) cholesterol concentration bears no relationship to the amount of fat in the diet (at least according to the SCS).
- Having a high blood cholesterol concentration may increase your risk of developing CHD.
- Having a high blood cholesterol concentration may reduce your risk of dying from a non-cardiac cause.
- Having a low blood cholesterol concentration may increase your risk of dying from a non-cardiac cause.
- Having either a high or a low blood cholesterol concentration will not influence how long you live.
Provided with this information, most sensible individuals would probably conclude that maybe the real message of the SCS was that it is best not to worry too much about your blood cholesterol concentration (except perhaps if it is low).
Ignored in all this evidence was the finding that age is the best predictor of CHD risk.
Since blood cholesterol concentrations increase with age (15, 35), is it not possible that blood cholesterol concentration is simply an indirect marker of increasing age and hence an increasing CHD risk?
Perhaps it was the sugar all along
Recall from the previous column (2) that Keys had clashed with, and soundly defeated his arch rival, John Yudkin, over the question of whether sugar rather than saturated fat was the real link between diet and CHD.
The problem for the SCS was that in 1999, its lead Italian researcher (24) reviewed the dietary data collected in the (rather small) sample of the original 12,770 participants (6, p. 42). Inconveniently, he discovered the correlation between “sweets” and coronary mortality was stronger than the correlation between animal products and coronary mortality. He subsequently confirmed “consumption of milk, butter, sugar, and pastries were ecologically related in a direct way with CHD mortality, whereas vegetable oils and alcohol did so in an inverse way” (31, p. 314). But “sugar” and “pastries” do not contain the same macronutrients as milk and butter, so how is this explained?
While arguing that “sugar” was not a better predictor of CHD risk than was saturated fat, Keys’ modern defenders (14) have conveniently ignored this finding related to “sweets” and “pastries.” It is not beyond the realms of possibility that if the SCS had properly measured diets in more than 4% of all participants in the trial, and if it had measured sugar intake more accurately, the study might have come to a different conclusion.
Instead, the SCS authors concluded “a diet low in animal and dairy products and rich in plant food may protect against coronary heart disease at the population level” (24).
Conveniently, these findings supported the messaging approved by their funders through the American Heart Association and National Heart, Blood, and Lung Institute.
Or perhaps it was the lack of physical activity?
One of the strongest predictors of future CHD in the SCS was the presence of Q waves on the electrocardiogram. This makes solid biological sense, for Q waves most often but do not always indicate that the patient has suffered a previous heart attack (myocardial infarction). So it makes sense that those who have suffered a previous heart attack must already have developed advanced coronary atherosclerosis that places them at increased risk for further heart attacks in the future.
In his original book about the SCS (7), Keys included a table showing the frequency of large Q waves in individuals in different populations, categorized by the amount of habitual physical activity undertaken (either light, moderate, or heavy). The bottom line of the table was the following:
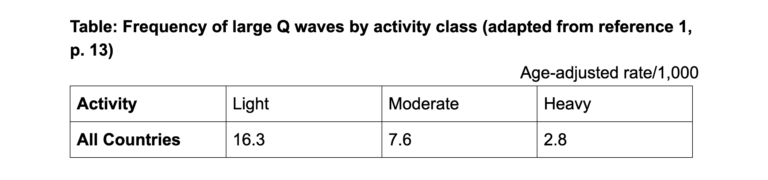
The table shows that in all countries, those in the light and moderate activity groups had a higher age-adjusted rate of Q waves than do those reporting participation in heavy exercise. This could suggest that those doing less exercise may have had more coronary atherosclerosis and were therefore at increased risk for future CHD.
As G. V. Mann explains, “The most sensible explanation for the national differences in CHD that they observed (in the SCS) was that exercise protects. That table mysteriously disappeared from the final publication” (1, p. 13).
Because the SCS was an observational study, it could never prove higher levels of exercise protected those reporting regular heavy exercise from the development of Q waves. Nor could it prove higher levels of physical activity rather than lower blood cholesterol concentrations could explain national differences in heart disease rates among participating subjects.
What the SCS could not disprove is that there is a collection of behaviors, more common in those who do more physical activity, that reduces the risk of developing CHD, regardless of individual blood cholesterol concentrations or dietary fat intake. Also, there are more individuals practicing heavy activity in countries with lower rates of CHD than in those with higher CHD rates.
Now that would be an interesting hypothesis to study.
Three subsequent studies failed to replicate key SCS findings
A scientific finding is only as good as subsequent replication shows it to be. In other words, until a study is replicated, its conclusions remain provisional, awaiting confirmation or refutation by subsequent studies that aim to study the same question, perhaps, as in this particular case, in relation to different populations.
There have now been four different publications that have addressed the key findings of the SCS in different populations.
1. Harcombe, whose Ph.D. thesis (39) showed there was no hard science to support the introduction of novel dietary guidelines in 1977 and 1984, has published two blog posts relevant to the findings of the SCS.
In the first (40), she evaluated the WHO data for cardiovascular death rates, all-cause death rates, and average blood cholesterol concentrations in 192 countries for which the data are available (41). When she plotted CHD deaths and deaths from all causes versus the average blood cholesterol concentrations in those 192 countries in both men and women, she found relatively weak inverse correlations with CHD deaths but much stronger inverse correlations with all-cause deaths, especially in women (Figure 3).
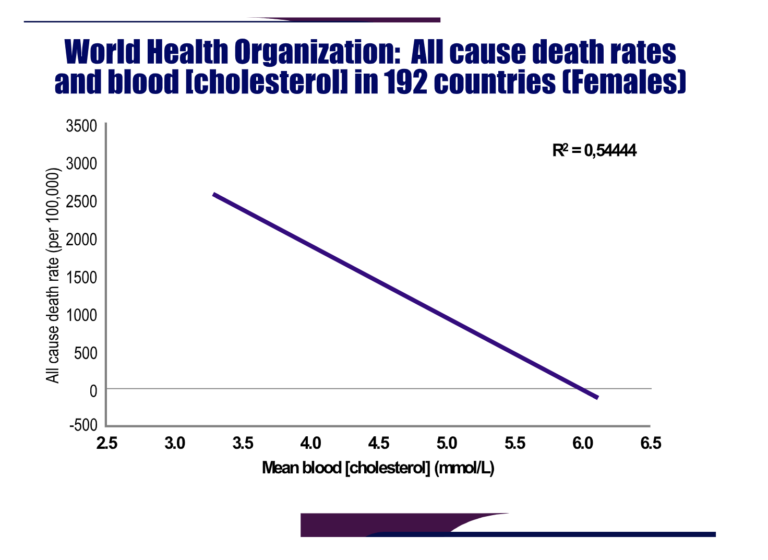
Figure 3: Harcombe plotted data sourced from the WHO website (41) to show all-cause death rates among women in 192 countries fell significantly as a function of increasing blood cholesterol concentrations. Reproduced from reference 40.
Thus, in this “arm chair” research, reminiscent of Keys’ methods in his original publication that ignited the fervor behind the diet-heart hypothesis, Harcombe was able to produce a completely opposite finding. What if these data had been available to Keys in 1953 (3) and 1957 (4)? What would he have made of them?
Harcombe next produced a very similar finding for CHD death rates and dietary energy percentages from saturated fat in a selection of European countries for which the data are available (42). Once again, the relationship was inverse: There was a linear but inverse relationship between dietary saturated fat and CHD deaths in both men and women. Again, the relationship was stronger in women. And once more, this “arm chair” research produced the opposite finding to that of Keys (3, 8). Harcombe’s research, using newer and more extensive information than was available to Keys, produced evidence that disproved both his conclusion (3, 8) and those of the SCS (23-25, 31).
2. In 2015, the Credit Suisse company issued a publication entitled “Fat: The New Health Paradigm” (43). The publication included a series of graphs showing relationships between dietary fats, both saturated and unsaturated, and CHD mortality rates in a number of European countries (Figures 4 and 5).
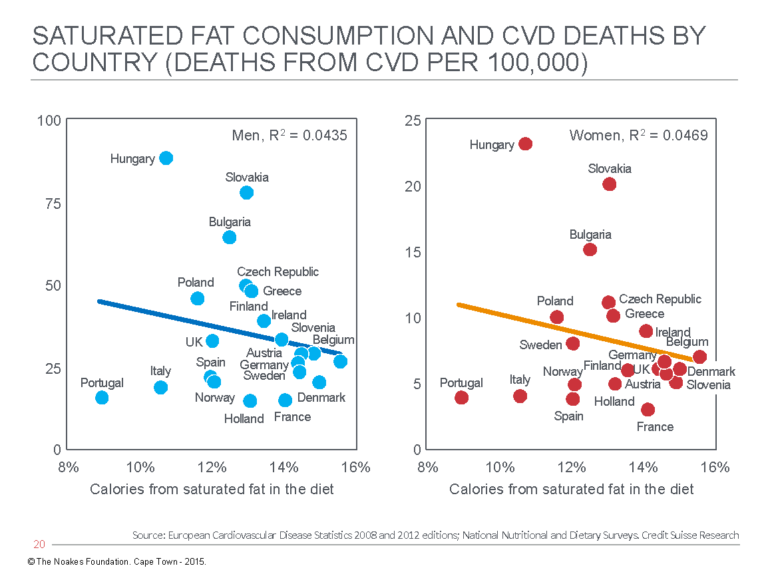
Figure 4: Cardiovascular death (CVD) rates in men (left panel) and women (right panel) in different European countries show an inverse association with the percentage of calories in the diet that are derived from saturated fat. Reproduced from reference 43.
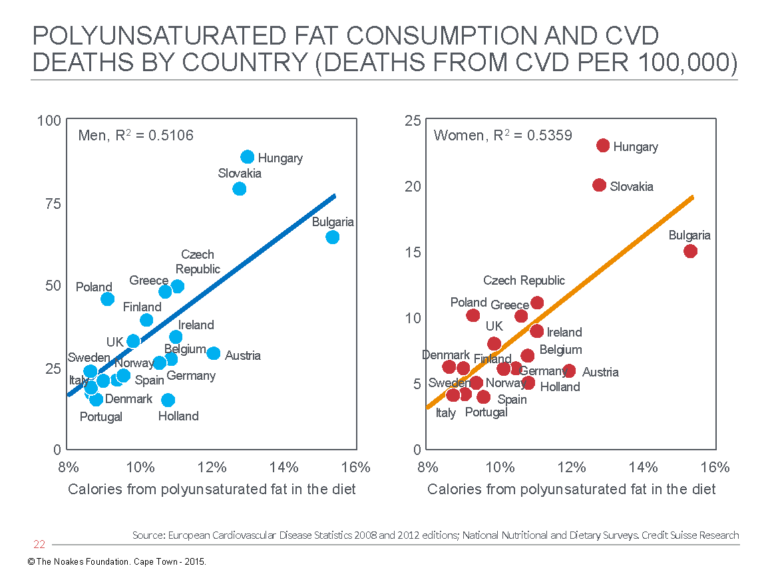
Figure 5: Cardiovascular death (CVD) rates in men (left panel) and women (right panel) in different European countries show a direct relationship with the percentage of calories in the diet that are derived from polyunsaturated fat. Reproduced from reference 43.
Figures 4 and 5 provide evidence that directly contradicts the findings of Keys and the SCS. In particular, the relationship between rising CVD death rates with increasing polyunsaturated fat intake is novel but in keeping with the real findings from Keys’ Minnesota Coronary Experiment, discussed subsequently.
3. In 2016, P. Grasgruber and colleagues (44) published an extensive analysis of the associations between food consumption and five CVD indicators in citizens in 42 European countries (Figure 6).
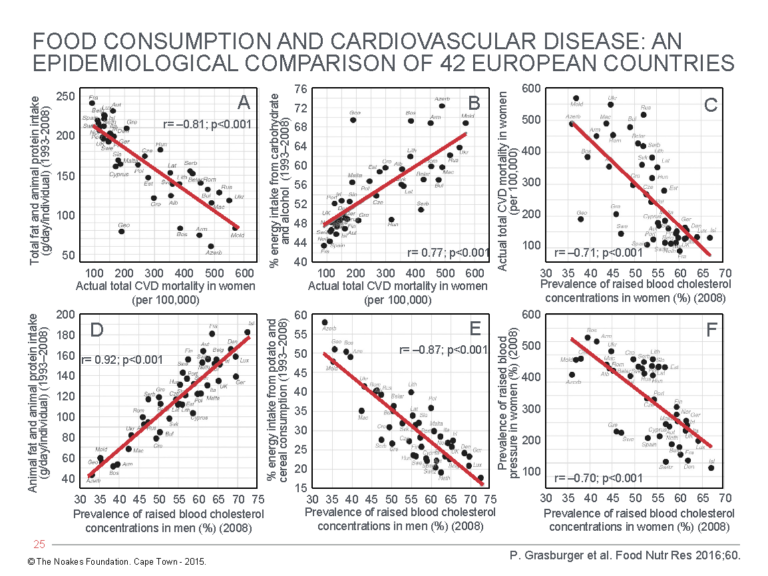
Figure 6: Associational relationships (A) between total fat and animal protein intake and total CVD mortality in women; (B) between % energy intake from carbohydrate and alcohol and total CVD mortality in women; (C) between total CVD mortality in women and prevalence of raised blood cholesterol concentrations; (D) between total animal fat and animal protein intake and prevalence of raised blood cholesterol concentrations in men; (E) between % energy intake from potato and cereal consumption and prevalence of raised blood cholesterol concentrations in men; and (F) between prevalence of raised blood pressure and prevalence of raised blood cholesterol concentrations in women. Reproduced from reference 44.
Their analysis showed:
- CVD mortality in both men and women rose as total fat and animal protein intake fell (Panel A). (Data for only the women are shown.)
- CVD mortality in both men and women rose with increasing carbohydrate and alcohol consumption (Panel B). (Data for only the women are shown.)
- CVD mortality in both men and women fell with an increasing prevalence of raised blood cholesterol concentrations (Panel C). (Data for only the women are shown.)
- Prevalence of raised blood cholesterol concentrations in men and women rose with increasing total fat and animal protein intake (Panel D). (Data for only the men are shown.)
- Prevalence of raised blood cholesterol concentrations in men and women fell with increasing potato and cereal consumption (Panel E). (Data for only the men are shown.)
- Prevalence of raised blood pressures in men and women fell with increasing prevalence of raised blood cholesterol concentrations (Panel F). (Data for only the women are shown.)
Any inferences drawn from this study would be opposite those coming from the SCS authors. The data collected by Grasgruber et al. suggest eating animal protein and fat is beneficial because it is associated with raised blood cholesterol concentrations and lowered blood pressures in both men and women. This is associated with reduced CVD mortality in both men and women.
In contrast, avoidance of animal fat and protein so more calories come from carbohydrates such as potatoes, cereals, and alcohol is associated with increased CV mortality, higher prevalence of high blood pressure, and lower blood cholesterol concentrations — the latter clearly a negative association from the results of this study.
Of course, the proviso remains that these relationships are purely associational and therefore cannot prove causation.
4. The Prospective Urban Rural Epidemiology (PURE) study, which began in January 2003, initially enrolled 148,723 individuals between 35 and 70 years old in 18 countries.
Following exclusions, the PURE study population was reduced to 135,335 individuals in whom daily dietary intake was carefully recorded (compared to just 499 in the SCS). The individuals were followed for a mean of 7.4 years, during which selected risk factors were measured at three, six, and nine years. During the study, the incidence of major cardiovascular events was recorded; there were 5,796 deaths, and another 4,784 individuals experienced a “major cardiovascular disease event.” The aim of the study was to assess the “association between consumption of carbohydrates, total fat, and each fat with cardiovascular disease and total mortality” (45, p. 2050). Some of the more important results of the study are shown in Figure 7.
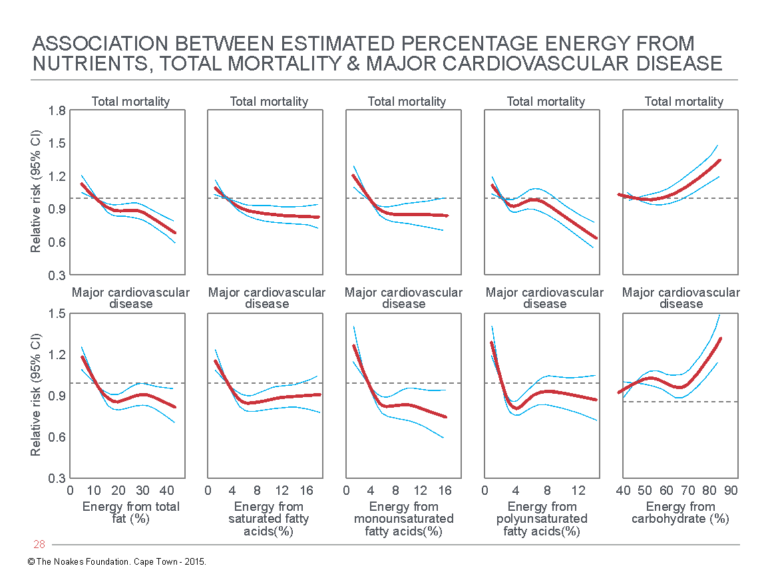
Figure 7: Relative risk for total mortality (top panels) and major cardiovascular disease (bottom panels) for: (Column 1) Increasing % energy from total fat; (Column 2) Increasing % energy from saturated fatty acids; (Column 3) Increasing % energy from monounsaturated fatty acids; (Column 4) Increasing % energy from polyunsaturated fatty acids; and (Column 5) Increasing % energy from carbohydrate. Reproduced from reference 45.
The clear evidence from Figure 7 is that whereas increasing energy intake from fat is associated with a progressive reduction in both total mortality and major cardiovascular disease, the opposite is the case for increasing energy from carbohydrates.
Again, these findings are the reverse of those from the SCS.
The PURE researchers have already published a further three reports (46-48), none of which contest this finding. Rather their finding on blood lipid differences associated with diets differing in their fat and carbohydrate contents are in line with the theme of this series of columns: They suggest increased carbohydrate intake may be associated with adverse changes in blood lipid concentrations, i.e., carbohydrate-induced hypertriglyceridemia (49).
Thus, the authors reported that the “intake of total fat and each type of fat was associated with higher concentrations of total cholesterol and LDL cholesterol, but also with higher HDL cholesterol and apolipoprotein A1 (ApoA1), and lower triglycerides, ratio of total cholesterol to HDL cholesterol, ratio of triglycerides to HDL cholesterol, and ratio of apolipoprotein B (ApoB) to ApoA1” (p. 774).
In contrast, “Higher carbohydrate intake was associated with lower total cholesterol, LDL cholesterol, and ApoB, but also with lower HDL cholesterol and ApoA1, and higher triglycerides, ratio of total cholesterol to HDL cholesterol, ratio of triglycerides to HDL cholesterol, and ApoB-to-ApoA1 ratio” (p. 774).
The authors concluded their data were “at odds with current recommendations to reduce total fat and saturated fats.” They explained:
Reducing saturated fatty acid intake and replacing it with carbohydrate has an adverse effect on blood lipids. Substituting saturated fatty acids with unsaturated fats might improve some risk markers, but might worsen others. Simulations suggest that ApoB-to-ApoA1 ratio probably provides the best overall indication of the effect of saturated fatty acids on cardiovascular disease risk among the markers tested. Focusing on a single lipid marker such as LDL cholesterol alone does not capture the net clinical effects of nutrients on cardiovascular risk. (p. 774)
The 1968-1973 Minnesota Coronary Experiment: Keys’ one and only substantive randomized controlLED trial
Although Keys would never acknowledge the flaws in the SCS study or the ways in which the study results challenged his hypotheses, he must have known that sooner or later he would have to venture into the unknown and design a study that would risk everything: a randomized controlled intervention trial (RCT) in which two matched groups would be studied. The one group would continue to eat the foods they normally ate; the other group would eat a diet in which their intake of saturated fat was reduced through replacement with a “healthier” fat — one that was known to reduce blood cholesterol concentrations, and preferably a vegetable oil full of omega-6 polyunsaturated fatty acids (PUFA) (50, 51). For his study, Keys chose the omega-6 PUFA linoleic acid. The dietary intervention also restricted the intake of dietary cholesterol to less than 150 mg daily.
The intervention Keys designed became known as the Minnesota Coronary Experiment (MCE). The study participants included 9,423 men and women between 20 and 97 years old who had been admitted to the either the Oak Terrace Nursing Home or one of six state mental hospitals. The study, funded by the National Heart Institute, exposed participants to either of the two diets (52, p. 4). In the intervention diet, liquid corn oil replaced the usual hospital cooking fats and was added to numerous other food items. Soft corn oil margarine was also used to replace butter. The control group continued to eat the standard hospital diet. The study lasted four and a half years.
Patients and investigators were all unaware of who received which diet. All meals for both diets were supplied on trays that appeared identical.
The dietary intervention successfully reduced the saturated fat intake by 50% (from 19 to 9% of total calories) and increased linoleic acid intake by more than 280% (from 3 to 13% of total calories). This diet reduced blood cholesterol concentrations by 14% compared to a 1% change in the control group.
Unlike the uncertainties produced in Keys’ associational studies, the design of the MCE was impeccable. If ever a study would provide a decisive answer, this would be it.
Interestingly, Keys set the percentage of fat in the diet in the MCE at 45%. This was the dietary fat content of the typical North American diet in the 1960s (53). Today, thanks to the influence of Keys and his various (failed) studies, the fat content percentage of the typical North American diet is substantially less, perhaps as low as 30% (53). This has been associated with a spectacular increase in body mass indices in U.S. men and women (Figure 8).
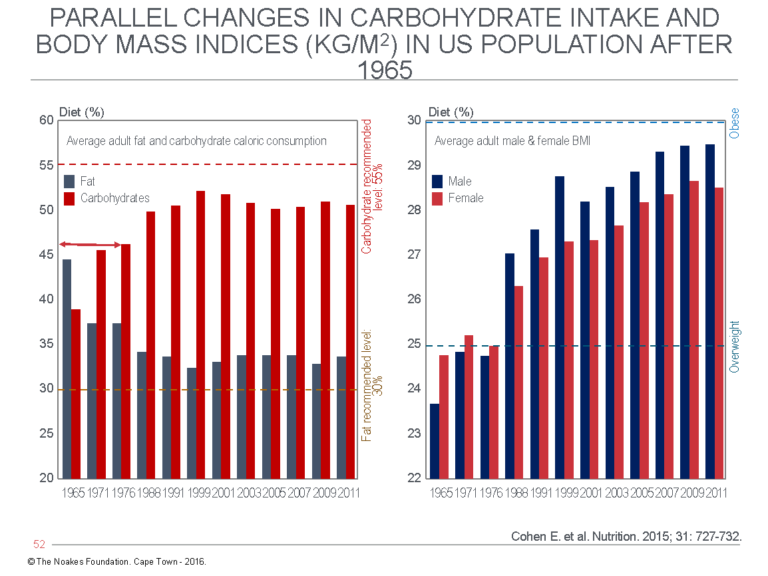
Figure 8: In 1965, 18 years after Keys first became interested in a dietary explanation for coronary heart disease, carbohydrates (red) provided 38% and fat 44% (blue) of daily calories in the average U.S. diet. By 2011, carbohydrate intake had increased to >50% and fat intake had decreased to ~34% (left panel). This has been associated with a progressive increase in the body mass indices of both U.S. men (blue) and women (red) (right panel). The dietary change was a direct result of the widespread, indeed global adoption of Keys’ (unproven) diet-heart and lipid hypotheses. Reproduced from reference 53.
The hypothesis tested in the MCE was as follows: If saturated fat causes CHD by increasing blood cholesterol concentrations, then its substitution in the diet by polyunsaturated fats (PUFAs) (which lower blood cholesterol concentrations) will reduce the number of cardiovascular events, including cardiac deaths, in the intervention group.
Remarkably, the first report of these hard endpoints (cardiovascular events including cardiac deaths) was published only in 1989 (54), although details of the trial methodologies and populations studied had been reported at a medical congress 14 years earlier (55, 56). Why was there such a long delay, given the importance of these findings for the formulation of the dietary advice that was even then in the process of being formulated by the U.S. Department of Agriculture (57)? What, indeed, would the 1977 McGovern commissioners have made of these findings? If the information had been available, might it have altered the commission’s final recommendation in 1977 (58)?
The 1989 MCE publication concluded: “The study did not show a statistically significant reduction in cardiovascular events or total deaths from the treatment diet” (54, p. 134, my emphasis).
This means the study neatly disproved both of Keys’ hypotheses.
It also means that in the course of normal science, Keys’ hypotheses should have been declared dead and buried in public in 1989, at the very latest. However, it seems this still is yet to happen in the majority of medical schools around the world.
Figure 9 shows that the dietary intervention did not influence the number of participants (as a percentage) who developed cardiovascular events during the 4.5 years of the trial (left panel), nor the percentage who died during the trial (right panel).
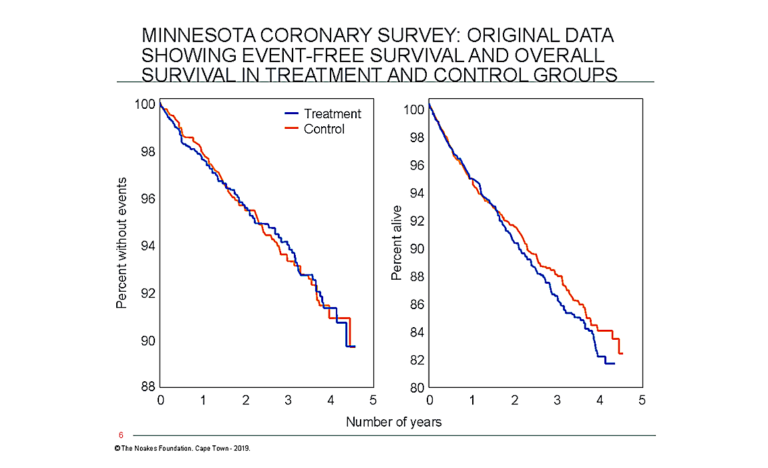
Figure 9: Although the results of Keys’ 1968-1973 MCE were available in the 1970s, the data on definitive medical outcomes were first published only in 1989 (54). These data showed there was no “statistically significant reduction in cardiovascular events or total deaths from the treatment diet” (54, p. 134). The panel on the left shows that the percentage of trial subjects who did not develop cardiovascular events during the 4.5 years of the study was not influenced by the intervention (blue line) compared to the control condition (orange line). The right panel shows that overall survival was also not influenced by the diet. Thus, this study disproved both of Keys’ hypotheses, the diet-heart and lipid hypotheses. Note that the percentage of participants still alive at 4.5 years (~82%) is substantially less than the percentage without events (~90%). This indicates significant mortality from diseases other than cardiovascular conditions, since cardiovascular events must include cardiac deaths. Reproduced from reference 54.
In other words, a dietary intervention (54) that specifically and effectively lowered blood cholesterol concentrations made no difference to the survival chances of those eating the low-cholesterol diet in which saturated fat was replaced with an increased consumption of the omega-6 PUFA linoleic acid.
In my book with Marika Sboros, Real Food on Trial (59, p. 78-79), we detail how Keys may actively have “buried” (60-62) the findings of the MCE, his very first RCT, when it became clear those findings disproved his hypotheses (52).
That is where the MCE story might have ended were it not for an unlikely discovery. In 2009, following the death of Dr. Ivan Frantz II, first author of the 1989 paper (54), Frantz’s son, Dr. Ivan Frantz III, found the boxes containing the original MCE data at their family home. This information was then re-analyzed, leading to another publication in 2016 (52), which reported findings that were somewhat different from those reported in the original 1989 publication (Figure 10).
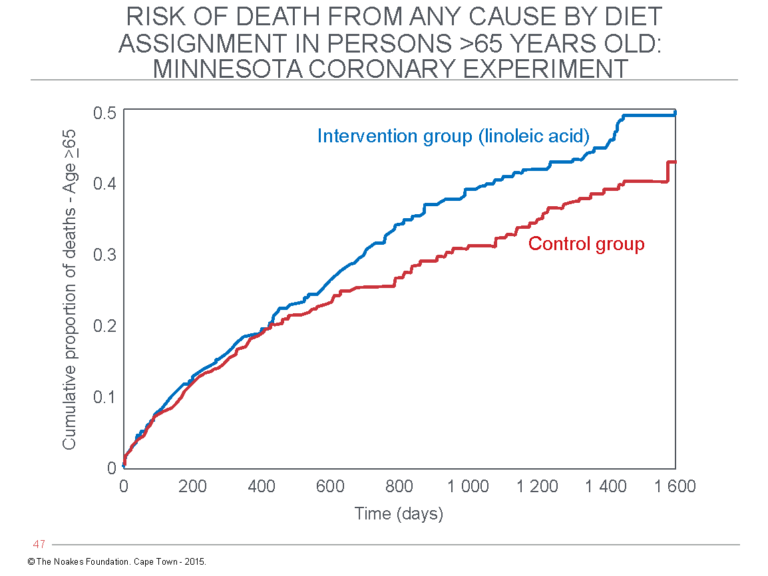
Figure 10: The “recovered” mortality data in the 1968-1973 MCE published in 2016 (52) found no evidence of cardiovascular benefit from the diet that lowered blood cholesterol concentrations. Instead, survival in participants over 65 years old receiving the intervention diet (blue line) was significantly worsened compared to the control group participants who continued to eat their usual diet (red line). Similar findings have been reported by the same authors for the recovered data from the very similar Sydney Diet Heart Study (63). The conclusion from these studies is that replacement of dietary saturated fat with the omega-6 PUFA linoleic acid may indeed lower blood cholesterol concentrations, but it provides no benefit and may indeed be harmful (52). This is not a message that is frequently heard. Reproduced from reference 52.
In particular, the recovered MCE mortality data found that the dietary intervention significantly reduced blood cholesterol concentrations but did not produce any benefits in terms of reduced overall mortality or fewer cardiovascular events, though such benefits had been reported in 1989. Instead, the new analysis found that the risk of death increased by 22% for every 30 mg/100 mL (0.78 mmol/L) reduction in blood cholesterol concentrations.
Older persons were the worst affected. Risk of death was increased by the dietary intervention in participants aged 65 or older (Figure 10).
It is inconceivable that Dr. Frantz II and the co-authors of the original MCE publication (54) were not aware of this finding. When they found there was no overall difference in mortality between groups, they most probably, according to Epstein’s logic, asked themselves this question: How are we going to spin this negative result? Surely there must be at least one sub-group of participants that benefited from the omega-6 PUFAs? Recall how Epstein had argued in 1977 that “an overall negative result for a trial as a whole does not rule out the possibility that intervention might be effective in such a sub-group” (33, p. 518-519).
If Frantz and his colleagues trolled through their data to see whether there was at least one sub-group that benefited from the intervention, during that process, they would have uncovered the sub-group of older males who were harmed by the intervention (Figure 10). This finding is especially important given that the elderly male sub-group is at the highest risk of having an adverse response because it contained more persons with advanced coronary artery disease. Thus, this group would be the first to show the adverse effects of a harmful diet — the classic “canary-in-the-cage” analogy from the mining industry.
Publication of those data shortly after they had been collected in 1974 would have destroyed Keys’ hypotheses and prevented the disastrous consequences, particularly the obesity epidemic (Figure 8; right panel), that were the direct result of the subsequent promotion and adoption of Keys’ low-fat, high-carbohydrate dietary ideas.
As the principal investigator of this trial, Keys would have known of these adverse findings already in 1974. He should have brought this information to the attention of the world, but he failed to act. We refer to this as negative evidence.
The concept of “negative evidence” comes from the episode in Sherlock Holmes’ story The Adventures of Silver Blaze. I referred to this case in my testimony before the Health Professions Council of South Africa (59, p. 265): “The fact that the dog did not bark when you would expect it to do so while a horse was stolen led Holmes to the conclusion that the evildoer was not a stranger to the dog, but someone the dog recognized and thus not cause him to bark. Holmes drew a conclusion from a fact (barking) that did not occur, which can be referred to as a ‘negative fact,’ or for the purposes of this discussion, an expected fact absent from the record” (64).
Summary
In this column, I reviewed Keys’ two most famous studies, the SCS and the MCE, the one an observational, associational study and the other one of the most thorough dietary RCTs yet completed.
The SCS could not find any evidence that diet was the exclusive determinant of the blood cholesterol concentration. It also established that having a higher or lower blood cholesterol concentration did not influence longevity, the really important measure of the efficacy of any medical intervention. The study did give partial support for Keys’ lipid hypothesis, finding that higher blood cholesterol concentrations were weakly linked to higher rates of CHD.
But given that (i) the study failed to establish that a high-fat diet is the most important cause of an elevated blood cholesterol concentration and (ii) that altering the blood cholesterol concentration was unlikely to influence longevity and all-cause mortality, the SCS could never be used to support a massive, population-based dietary change.
Appreciating this, Keys and his followers successfully spun the findings as if they confirmed his hypotheses (32, 33).
The MCE, on the other hand, utterly destroyed both of Keys’ hypotheses by showing that a dietary change that reduced the intake of saturated fat by replacing it with an omega-6 PUFA linoleic acid — an intervention that successfully lowered the blood cholesterol concentration — increased CHD rates in the intervention group (Figure 10).
The moment those data became available, Keys’ hypotheses should have been dead and buried.
Knowing that, Keys had two choices: Either acknowledge both hypotheses were wrong or obscure the data.
He chose the latter.
And he was successful until, inconveniently for his reputation and how he will be remembered, they were discovered and published in 2016 (52), 12 years after his death at age 100.
But his death does not absolve his guilt.
ADDITIONAL READING
- It’s the Insulin Resistance, Stupid: Part 1
- It’s the Insulin Resistance, Stupid: Part 2
- It’s the Insulin Resistance, Stupid: Part 3
- It’s the Insulin Resistance, Stupid: Part 4
- It’s the Insulin Resistance, Stupid: Part 5
- It’s the Insulin Resistance, Stupid: Part 6
- It’s the Insulin Resistance, Stupid: Part 7

Professor T.D. Noakes (OMS, MBChB, MD, D.Sc., Ph.D.[hc], FACSM, [hon] FFSEM UK, [hon] FFSEM Ire) studied at the University of Cape Town (UCT), obtaining a MBChB degree and an MD and DSc (Med) in Exercise Science. He is now an Emeritus Professor at UCT, following his retirement from the Research Unit of Exercise Science and Sports Medicine. In 1995, he was a co-founder of the now-prestigious Sports Science Institute of South Africa (SSISA). He has been rated an A1 scientist by the National Research Foundation of SA (NRF) for a second five-year term. In 2008, he received the Order of Mapungubwe, Silver, from the President of South Africa for his “excellent contribution in the field of sports and the science of physical exercise.”
Noakes has published more than 750 scientific books and articles. He has been cited more than 16,000 times in scientific literature and has an H-index of 71. He has won numerous awards over the years and made himself available on many editorial boards. He has authored many books, including Lore of Running (4th Edition), considered to be the “bible” for runners; his autobiography, Challenging Beliefs: Memoirs of a Career; Waterlogged: The Serious Problem of Overhydration in Endurance Sports (in 2012); and The Real Meal Revolution (in 2013).
Following the publication of the best-selling The Real Meal Revolution, he founded The Noakes Foundation, the focus of which is to support high quality research of the low-carbohydrate, high-fat diet, especially for those with insulin resistance.
He is highly acclaimed in his field and, at age 67, still is physically active, taking part in races up to 21 km as well as regular CrossFit training.
References
- Mann GV. A short history of the diet/heart hypothesis. In: Coronary Heart Disease. The dietary sense and nonsense. An evaluation by scientists. Mann GV, Ed., London, England: Janus Publishing Company, 1993. 1-17.
- Noakes TD. It’s the insulin resistance, stupid: Part 6. CrossFit.com. 30 October 2019. Available here.
- Keys A. Atherosclerosis: A problem in newer public health. J Mt Sinai Hosp NY 20(1953): 118-139.
- Keys A, Grande F. Role of dietary fat in human nutrition. III – Diet and the epidemiology of coronary heart disease. Am J Pub Health 47(1957): 1520-1530.
- Yerushalmy J, Hilleboe HE. Fat in the diet and mortality from heart disease. A Methodological note. New Y State J Med. 57(1957): 2343-2354.
- Teicholz N. The Big Fat Surprise: Why Butter, Meat and Cheese Belong in a Healthy Diet. New York, NY: Simon and Schuster, 2014.
- Keys A. Seven Countries: A Multivariate Analysis of Death and Coronary Heart Disease. Cambridge, MA: Harvard University Press, 1980.
- Melicow MM. Percival Pott (1713–1788): 200th anniversary of first report of occupation-induced cancer of scrotum in chimney sweepers (1775) Urology 6(1975): 745.
- Doll R, Hill AB. Smoking and carcinoma of the lung. Preliminary report. BMJ 2(1950): 739-748; Petro R, Darby S, Deo H, et al. Smoking, smoking cessation, and lung cancer in the UK since 1950: combination of national statistics with two case-control studies. BMJ 321(2000): 323-329.
- Snow J. On the mode of communication of cholera. Second Edition; John Churchill, London; New Burlington Street. 1854; Vandenbroucke JP, Eelkman Rooda HM, Beukers H. Who made John Snow a hero? Am J Epidemiol. 133(1991): 967-973.
- Hill AB. The environment and disease: association or causation? Proc R Soc Med. 58(1965): 295-300; Lucas RM, McMichael AJ. Association or causation: Evaluating links between “environment and disease”. Bull WHO 83(2005): 792-795.
- Ioannidis JPA. Implausible results in human nutrition research. Definitive solutions won’t come from another million observational papers or small randomized trials. BMJ 347(2013): f6698; Brown AW, Ioannidis JPA, Bier DM, et al. Unscientific beliefs about scientific topics in nutrition. Adv Nutr 5(2014): 563-565; Ioannidis JPA. We need more randomized trials in nutrition – preferably large, long-term, and with negative results. Am J Clin Nutr. 103(2016): 1385-1386; Ioannidis JPA. Viewpoint: The challenge of reforming nutritional epidemiologic research. JAMA 320(2018): 969-97.
- Wood PDP. A possible selection effect in medical science. J Roy Stat Soc. 30(1981): 131-135.
- Pett KD, Kahn J, Willett WC, et al. Ancel Keys and the Seven Countries Study: An evidence-based response to revisionist histories. White Paper commissioned by the True Health Initiative. 1 August 2017. Available here.
- Sekikawa A, Miyamato Y, Miura K, et al. Continuous decline in mortality for coronary heart disease in Japan despite a continuous and marked rise in total cholesterol: Japanese experience after the Seven Countries Study. Int J Epidemiol. 44(2015): 1614-1624.
- Murata M. Secular trends in growth and changes in eating patterns of Japanese children. Am J Clin Nutr. 72(2000): 1379S-1383S.
- Guberan E. Surprising decline of cardiovascular mortality in Switzerland: 1951-1976. J Epidemiol Comm Health. 33(1979): 114-120.
- Keys A. Epidemiological studies related to coronary heart disease: Characteristics of men aged 40-59 in seven countries. Acta Med Scand. 460(1966): 1-392.
- Keys A, Aravanis C, Van Buchem FSP et al. The diet and all-causes death rate in the Seven Countries Study Lancet 318(1981): 58-61.
- Keys A, Menotti A, Aravanis C, et al. The Seven Countries Study: 2,289 deaths in 15 years. Prev Med. 13(1984): 141-154.
- Keys A, Menotti A, Karvonen MJ, et al. The diet and 15-year death rate in the Seven Countries Study. Am J Epidemiol. 124(1986): 903-915.
- Menotti A, Keys A, Aravanis C, et al. Seven Countries Study. First 20-year mortality data in 12 cohorts of six countries. Ann Med. 21(1988): 175-179.
- Menotti A, Keys A, Kromhout D, et al. Inter-cohort differences in coronary heart disease mortality in the 25-year follow-up of the Seven Countries Study. Europ J Epidemiol. 9(1993): 527-536.
- Menotti A, Kromhout D, Blackburn H, et al. Food intake patterns and 25-years mortality for coronary heart disease: Cross-cultural correlations in the Seven Countries Study. Europ J Epidemiol. 15(1999): 507-515.
- Blackburn H, Menotti A. Major results of the Seven Countries Study. In: Kromhout D, Menotti A, Blackburn H. The Seven Countries Study. A scientific adventure in cardiovascular disease epidemiology. Bilthoven, Netherlands: Marjan Nijssen-Kramer, Studio RIVM, 1993. pp.159-174.
- Noakes TD. 1996 JB Wolffe Memorial Lecture. Challenging beliefs: ex Africa semper aliquid novi. Med Sci Sports Exerc. 29(1977): 571-590.
- Levenstein H. Fear of Food: A History of Why We Worry About What We Eat. London, U.K.: University of Chicago Press, 2012.
- Noakes TD. It’s the insulin resistance, stupid! Part 10. (forthcoming)
- Keys A. Sucrose in the diet and coronary heart disease. Atherosclerosis 14(1971): 193-202.
- Keys A, Grande F, Anderson JT. Bias and misrepresentation revisited: Perspective on saturated fat. Am J Clin Nutr. 27(1974): 188-212.
- Menotti A, Puddu PE. Can we still learn from the Seven Countries Study? Curr Op Lipidol. 29(2018): 313-317.
- Epstein FH. Public health implications of the Seven Countries Study. In: Kromhout D, Menotti A, Blackburn H. The Seven Countries Study. A scientific adventure in cardiovascular disease epidemiology. Bilthoven, Netherlands: Marjan Nijssen-Kramer, Studio RIVM, 1993. pp. 169-174.
- Epstein FH. Preventive trials and the “diet-heart” question: Wait for results or act now? Atherosclerosis 26(1977): 515-523.
- The Coronary Drug Project Research Group: Clofibrate and niacin in coronary heart disease. JAMA 231(1975): 360.
- Kannel WB. Metabolic risk factors for coronary heart disease in women: Perspective from the Framingham Study. Am Heart J. 154(1987): 413-419.
- Kearns CE, Schmidt LA, Glantz SA. Sugar industry and heart disease research. A historical analysis of internal industry documents. JAMA Internal Medicine 176(2016): 1680-1685.
- Fidanzi F, Alberti A, Lanti M, et al. Mediterranean Adequacy Index: correlation with 25-year mortality from coronary heart disease in the Seven Countries Study. Nutr Metab Cardiovasc Dis. 14(2004): 254-258.
- Harcombe Z, Baker JS, Davies B. Food for thought: Have we been giving the dietary advice? Food Nutr Sci. 4(2013): 240-244.
- Harcombe Z. An examination of the randomized controlled trials and epidemiological evidence for the introduction of dietary fat recommendations in 1977 and 1984: A systematic review and meta-analysis. Ph.D. thesis, University of West Scotland, March 2016.
- Harcombe Z. Blogpost: Cholesterol and heart disease – There is a relationship, but it’s not what you think. 23 November 2010. Available here.
- World Health Organisation. Stats. Global Health Observatory data repository. Cholesterol. Available here.
- Harcombe Z. Blogpost: Saturated fat and CHD in Europe. 1 March 2010. Available here.
- Credit Suisse Research Institute. Fat: The New Health Paradigm. September 2015. Available here.
- Grasgruber P, Sebera M, Hrazdira E, et al. Food consumption and the actual statistics of cardiovascular diseases: an epidemiological comparison of 42 European countries. Food Nutr Res. 60(2016): 31694.
- Dehghan M, Mente A, Zhang X, et al. Association of fats and carbohydrate intake with cardiovascular disease and mortality in 18 countries from five continents (PURE): A prospective cohort study. Lancet 390(2017): 2050-2062.
- Mente A, Dehghan M, Rangarajan S, et al. Association of dietary nutrients with blood lipids and blood pressure in 18 countries: A cross-sectional analysis from the PURE study. Lancet 390(2017): 774-787.
- Miller V, Mente A, Dehghan M, et al. Fruit, vegetable, and legume intake, and cardiovascular disease and deaths in 18 countries (PURE): A prospective cohort study. Lancet 390(2017): 2037-2049.
- Dehghan M, Mente A, Rangarajan S, et al. Association of dairy intake with cardiovascular disease and mortality in 21 countries from five countries (PURE): A prospective cohort study. Lancet 392(2018): 2288-2297.
- Noakes TD. It’s the insulin resistance, stupid: Part 1. CrossFit.com. 6 July 2019. Available here.
- Keys A, Anderson JT, Grande F. Serum cholesterol response to change in the diet: IV. Particular saturated fatty acids in the diet. Metabolism 14(1965): 776-787.
- Anderson JT, Grande F, Keys A. Cholesterol-lowering diets. J Am Dietet Assoc 62(1973): 133-142.
- Ramsden CE, Zamora D, Majchrzak-Hong S, et al. Re-evaluation of the traditional diet-heart hypothesis: analysis of recovered data from Minnesota Coronary Experiment (1968-73). BMJ 353(2016): i1246.
- Cohen E, Cragg M, deFonseka J, et al. Statistical review of US macronutrient consumption data, 1965–2011: Americans have been following dietary guidelines, coincident with the rise in obesity. Nutrition 31(2015): 727-732.
- Frantz ID, Dawson EA, Ashman PL, et al. Test of effect of lipid lowering by diet on cardiovascular risk. The Minnesota Coronary Survey. Arteriosclerosis 9(1989): 129-135.
- Brewer ER, Ashman PL, Kuba K. The Minnesota Coronary Survey: composition of their diets, adherence, and serum lipid response. Circulation 51.suppl 2(1975): 1-269.
- Dawson E, Gatewood L. The Minnesota coronary survey: methodology and characteristics of the population. Circulation 52.suppl 2(1975): 271.
- 2015 Dietary Guidelines Advisory Committee. History of Dietary Guidance Development in the United States and the Dietary Guidelines for Americans. Available here.
- U.S. Senate Select Committee on Nutrition and Human Needs. Dietary Goals for the United States, 2nd ed. Washington, DC, U.S. Government Printing Office, 1977. (See also https://health.gov/dietaryguidelines/dga2005/report/PDF/G5_History.pdf).
- Noakes TD, Sboros M. Real Food on Trial. How the Diet Dictators Tried to Destroy a Top Scientist. U.K.: Columbus Publishing Ltd., 2019.
- Begley S. Records found in dusty basement undermine decades of dietary advice. Raw data from a 40-year-old study raises new questions about fats. Sci Amer. 19 April 2017.
- O’Connor A. A decades-old study, rediscovered, challenges advice on saturated fat. The New York Times. 13 April 2016.
- Whoriskey P. This study 40 years ago could have reshaped the American diet. But it was never published. The Washington Post. 12 April 2016.
- Ramsden CE, Zamora D, Leelarthaepin B, et al. Use of dietary linoleic acid for secondary prevention of coronary heart disease and death. Evaluation of recovered data from the Sydney Diet Heart Study and updated meta-analysis. BMJ 346(Feb. 4, 2013): e8707.
- Skotnicki M. “The Dog that Didn’t Bark:” What we can learn from Sir Arthur Conan Doyle about using the absence of expected facts. Briefly writing. 25 July 2012. Available here.
Comments on It’s the Insulin Resistance, Stupid: Part 8
http://www.survivediabetes.com/Capture.JPG
Both 100 ...
Very nicely done - that Cohen study is a telling graphic for the obesity epidemic and the MCE material is damning.
I'm a psychotherapist, and Keys exhibited almost all the traits of narcisistic personality disorder: self-importance, entitlement, exaggerates achievements, belittle people they perceive as inferior, take advantage of others, unwilling to recognize the needs and feelings of others, arrogant or haughty - https://www.mayoclinic.org/diseases-conditions/narcissistic-personality-disorder/symptoms-causes/syc-20366662 which suggest to me he may have been more interested in acclaim than whether his diet actually worked or not.
I have a pic of Keys at 100 in San Diego, at his last public appearance. He is obese, the asymmetry of his face suggests a stroke, and he is in a wheelchair. I have a pic of a Kitavan male, believed also to be 100, who is obviously in good health. Interestingly, the Kitavans pretty much eat Keys's low fat diet. Makes me wonder about the refined carbohydrates in the Western diet. I couldn't find a way to past these pics here but I could email it to you in case you haven't seen it jonty2@earthlink.net
PS Comments aren't working properly, says thre are 3 but no comments are visible.
What role is liver bioaccumulation of persistent nanoparticulate aluminum from vaccine adjuvants playing in displipidemia and subsequent health outcomes?
“Mitochondrial metabolism is the main site of the toxicological action of Al.”
Hepatic response to aluminum toxicity: dyslipidemia and liver diseases.
Mailloux RJ1, Lemire J, Appanna VD.
https://www.ncbi.nlm.nih.gov/pubmed?term=hepatic%20response%20to%20aluminum%20toxicity%3A%20dyslipidemia%20and%20liver%
I know its just words on a computer screen, but such texts make me rage. Unbelievable.
Then I checked the current medications in my home country (Switzerland). Seemingly in the year 2018 there have been 775 mio spent by 2 mio people (out of a small population of 8 mio) for medications that, accordingly to that post, elevate overall death risk.
Medications paid within “one of the best health care system on earth”.
Cardio related medicine is on place 4. 1st place in medications is against cancer, 3rd place for metabolism.
That’s why I do Crossfit.
Finland was the pinnacle of the Keys' study, i.e. the worst case. Well, ca. 200 hard working forest workers in Karelia represented it. Seems that Finlands gene pool is divided into two; more sickness in the east (russian influence?) than in the west (swedish?). North-karelia is in the east, and their diet was heavy in full milk, cheese etc.
Keys made friends there, actually retiring to Italy together with one of those, and he had great influence on affairs. The resulting massive margarine campaign, North-Karelia project, has been cited frequently, but also analyzed by Uffe Ravnskov. The dogma prevails strongest in Finland, I would presume.
An example of Nutrion authorities Q&A about weaning, sure a topic of Professor Noakes' expertise:
"Q: Need for an extra fat intake, when a child drinks skim milk: “Eat together 2018” - the recommendation does not any more mention additional fat. Can the fat intake of young children be too low?
Answer: A separate recommendation for using additional fat was no longer considered necessary based on the results of the STRIP study*. Children's fat intake is sufficient. The new recommendation has therefore removed the separate "milkfat-dependent" advice on additional fat for 1-2-year-old children.
Recommended intake to all children from the age of one year (when weaning from breast milk or infant formula to skim / low-fat milk) is 20-30 g of fat (1.5 to 2 tablespoons of seed oil or 4-6 teaspoons of> 60% vegetable oil margarine). Soft vegetable fats, preferably in form of oil**, ensure the adequate supply of essential fatty acids to infants.
* they promoted seed oil use, and there was a control group “fare as normally”. Keys style trial and horror. Long follow-up until teen age, and now this is a recommendation.
** local canola oil aka rapeseed oil likely. "
So, growing up means giving up hi-fat breast milk for skim milk + seed oil. Luckily the sugared milk drinks are mostly absent, but skim milk is the milk of choice at kindergartens. Healthy, science based advice? So they say.
JR
It’s the Insulin Resistance, Stupid: Part 8
5