
When medical scientists propagate a false hypothesis, two things happen, and both of them are bad.
First, the wrong idea causes direct harm to those who adopt practices based on that incorrect hypothesis. Second, the wrong idea suppresses any attempts to discover the correct hypothesis. Such suppression occurs as a result of (enforced) scientific consensus.
Anyone who dares to question the false but agreed-upon hypothesis is labeled a “hypothesis skeptic” or “hypothesis denier.” Very soon, that individual finds herself a scientific pariah, shunned and publicly humiliated by her colleagues, no longer able to secure research funding. In this way skeptics are conveniently and very effectively removed from the scientific mainstream. This technique is now recognized as academic mobbing (2) and ritual degradation (3). The consequences for the victim of academic mobbing and ritual degradation are usually calamitous.
Having personally traversed this academic minefield for the past nine years, I understand it rather too well (4).
But the reality is that science is never settled, and skeptics will always play a crucial role in driving scientific progress.
Ancel Keys’ incorrect diet-heart hypothesis that saturated fat is the direct cause of heart attacks and death from coronary heart disease (CHD) led directly to its offspring, the lipid hypothesis, which holds that an elevated blood cholesterol concentration is the singular cause of CHD. This in turn led to a multibillion-dollar industry focused on reducing blood cholesterol concentrations, principally through the prescription of cholesterol-lowering statins and “aided” by a low-fat, high-carbohydrate (LFHC) diet. This diet recommendation was enshrined in the 1977 U.S. Dietary Guidelines for Americans (USDGA) (5).
The 1977 USDGA and other forms of continued support for the diet-heart and lipid hypotheses have led to at least three dire consequences (4).
First, the incidences of obesity and Type 2 diabetes mellitus (T2DM) have more than doubled in this period (6). The reasons for this will be explained. Importantly, the key pathological feature of T2DM is widespread progressive obstructive arterial disease in all the major arterial systems in the body but especially the arteries supplying the kidneys and lower limbs — thus the growing global pandemic of kidney failure and lower-limb amputations.
As a result of the worldwide adoption of the USDGA based on Keys’ false diet-heart hypothesis, the LFHC diet has been promoted as the ultimate intervention to prevent obstruction of the coronary arteries. Yet, very inconveniently, the promotion of this diet has clearly produced a much more devastating outcome — the worst possible forms of obstructive arterial disease in persons with T2DM.
Second, despite the almost universal prescription of statin drugs to anyone considered at even the slightest risk of ever developing CHD, after five or more decades in decline, the global incidence of CHD has begun to increase in some countries (7). Clearly, the billion-dollar statin drug industry that thrives by promising to prevent all future heart attacks is not delivering on its puffery. Evidence of the ineffectiveness of statins is perhaps the final repudiation of both the diet-heart and lipid hypotheses.
Third, because these two hypotheses were embraced so fanatically (and without proper due scientific process), any attempts by skeptics to develop alternate hypotheses have been rigorously suppressed in part by labeling the challengers as “cholesterol-skeptics” or “cholesterol-denialists” (8).
The ultimate tragedy is that the one theory that best explains why the adoption of the LFHC diet has destroyed global health has been ignored. It is not taught in even a minority of medical schools around the world. This theory holds that a single biological state, insulin resistance syndrome (IRS), is the key driver of most of the chronic medical conditions to which modern humans fall prey. This theory is the work of a single researcher, Dr. Gerald Reaven, recently deceased, and his small team of researchers at Stanford University in Palo Alto, California.
It is my argument that Reaven’s work is perhaps the most important body of medical research of the last five decades. His work is so far ahead of current medical thinking that, sadly, Reaven died before his work’s value was properly recognized with a Nobel Prize. But his time of recognition will come. The moment is rapidly approaching when the medical profession will be forced to admit the genius in Reaven’s work. The truth cannot be denied forever.
The Discovery of Insulin Resistance Syndrome
Reaven spent 60 years describing the condition that would become his trademark, insulin resistance syndrome (IRS).
His academic interest (1, 9) was stirred early in his career when he read the work of Harry Himsworth (10), who already in the 1930s had proposed that there are two forms of diabetes. The first, insulin-deficient Type 1 diabetes mellitus (T1DM), is caused when the pancreatic insulin-secreting beta cells are destroyed by an autoimmune process of unknown origin. As a result, the affected person is left without any ability to produce insulin. Such persons cannot live without regular insulin injections.
It was this group of patients whose lives were so dramatically changed with the discovery of insulin by Frederick Banting, John Macleod, Charles Best, and James Collip in December 1921 (11). Because insulin is present in the blood in such small amounts, at the time of insulin’s discovery, it was not possible to accurately measure blood insulin concentrations. (Banting and Macleod won the Nobel Prize for isolating a pancreatic substance that reduced blood insulin concentrations in those with T1DM. At the time, they knew only that insulin was a protein present in pancreatic tissue. The structure of insulin was first characterized by another Nobel Prize winner, Dorothy Hodgkin, in 1968 (12)). The natural assumption, then, was that all forms of diabetes are caused by the same mechanism: an absolute deficiency in circulating blood insulin concentrations, as found in T1DM.
But Himsworth came up with a different explanation, seemingly from nowhere (10). He understood that in all forms of diabetes, the tissues have a reduced capacity to take up glucose. He did not agree, however, that this was always due to the complete absence of the hormone insulin, one action of which is to promote glucose uptake by the tissues, particularly the liver, heart, and skeletal muscles.
Instead, he proposed, “The diminished ability of the tissues to utilize glucose is referable either to a deficiency of insulin or to insensitivity to insulin, although it is possible that both factors may operate simultaneously.” Accordingly, he argued that diabetes should be subdivided into two categories: “insulin-sensitive and insulin-insensitive types.” He also noted that there were clear differences in the clinical expression of these two subtypes so that “insulin-sensitive diabetes, which is thought to be due to a deficiency of insulin, tends to be severe … whereas diabetes due not a lack of insulin but to insensitivity to insulin, is generally less severe.”
So by 1949, Himsworth had concluded, “It appears we should accustom ourselves to the idea that a primary deficiency of insulin is only one, and then not the commonest, cause of the diabetes syndrome” (13). It would take another 40 years before the National Diabetes Data Group would formally acknowledge this distinction (14).
Today, we understand that in persons with IRS, especially those who ultimately develop T2DM, the target cells on which insulin normally acts, especially those in the pancreas and liver but also in many other organs, become progressively more resistant to the normal action of insulin over years and even decades. As a result, insulin must be secreted in increasingly greater amounts, producing the progressive IRS that Reaven’s methodical research ultimately discovered.
But after perhaps two to three decades of this daily need to oversecrete insulin, the pancreatic beta cells become exhausted; the pancreas fails; blood insulin concentrations fall; and the patient develops the characteristic features of T2DM, including very high blood glucose concentrations with the appearance of glucose in the urine.
Reaven demonstrated that most persons with IRS do not develop T2DM. However, this does not mean those who have IRS that does not progress to T2DM will live long and disease-free lives.
In fact, Reaven’s unique contribution has been to show that IRS is the precursor for essentially all the chronic medical conditions that currently plague modern humans, from acne and Alzheimer’s disease or dementia to hypertension, peripheral vascular disease, polycystic ovarian syndrome, coronary heart disease, and perhaps even cancer.
Thus, while we fret over the growing pandemic of T2DM, we need to understand that this is only the tip of the disease iceberg; hidden underneath lies an even greater epidemic of chronic modern diseases caused by IRS in those who will not ever develop T2DM but who will nevertheless suffer from any number of a wide array of conditions that, in our ignorance, we continue to call “diseases of lifestyle.” These diseases, as I will show, are more correctly termed “diseases of the modern industrial diet” of highly processed foods.
Since IRS is the key driver of elevated blood pressure (hypertension) (15) and coronary artery disease (16), in his repudiation of the simplistic diet-heart and lipid hypotheses, Reaven also established that “coronary heart disease risk factors in normotensive, nondiabetic individuals includes more than a high LDL cholesterol concentration” (1).
To fully comprehend the nature of modern human ill health, we need first to understand the IRS.
Reaven Becomes Interested in Insulin Insensitivity
Today, it is rather easier to distinguish between T1DM, T2DM, and IRS in affected patients. All one need do is measure blood insulin concentrations. If endogenous (produced by the patient’s body) insulin is absent, the patient has T1DM; if insulin is present, the patient may have either T2DM or IRS.
But when Reaven began his work, the ability to effectively measure blood insulin concentrations was still new. It had only just been achieved by Rosalyn Yalow and Solomon Berson in 1960. Working at the Veterans Administration (VA) hospital in the Bronx, New York, Yalow and Berson developed an immunoassay method to accurately measure the tiny amounts of insulin in the blood (17). For this, Yalow was awarded the Nobel Prize in 1977.
Yalow and Berson wasted no time in showing that blood insulin concentrations were, on average, higher in persons with T2DM than in healthy subjects without the disease. They concluded: “The tissues of the maturity-onset diabetic do not respond to his insulin as well as the tissues of the nondiabetic subject respond to his insulin” (16). Thus, they confirmed Himsworth’s postulate from two decades earlier: that those with T2DM are “insulin insensitive.”
Yet, no one at that time understood exactly what constitutes “insulin insensitivity.” Reaven would devote the remainder of his working life to the explanation of this phenomenon.
Reaven’s Interest Piqued by Elevated Blood Triglyceride Concentrations
Reaven began his research in the 1960s, at a time when Keys’ diet-heart and lipid hypotheses were gaining great traction globally. Reaven, (like most medical doctors around the world then and now) was taught that an elevated blood cholesterol concentration “was considered the primary culprit in heart disease” (18, p. 47).
But what was Reaven to make of Margaret Albrink and Evelyn Man’s 1959 findings (19), which showed blood cholesterol concentrations appeared to be no higher in those who had suffered heart attacks (many of whom had T2DM) than they were in normal patients without established heart disease (Figure 1)?
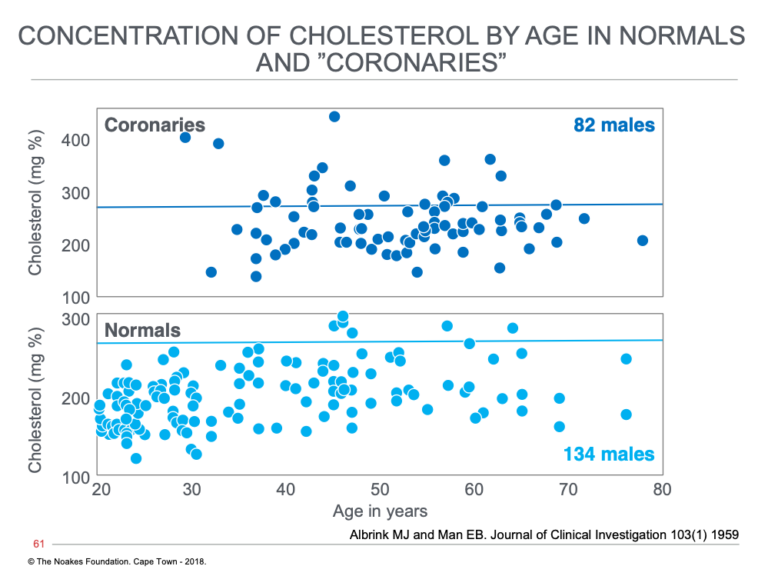
Figure 1: The distribution at different ages of blood cholesterol concentrations in persons without (normals) and those with diagnosed heart attack (acute myocardial infarction) (coronaries). Note that the majority of coronaries have what were then considered normal blood cholesterol concentrations (below horizontal line at a cholesterol concentration of 280 mg%). Reproduced from reference 19.
The usual response to such information is to ignore it, to pretend that it does not exist, as indeed has been the common practice for the past six decades. But clearly Reaven was made of sterner stuff. He knew a paradox when he saw one, and his personality was such that the uncertainty revealed by this paradox would drive him to examine that enigma until its truth was exposed.
Albrink and Man also reported that blood triglyceride concentrations were different in normal and coronary groups, and appeared to be higher in those with established heart disease (Figure 2).
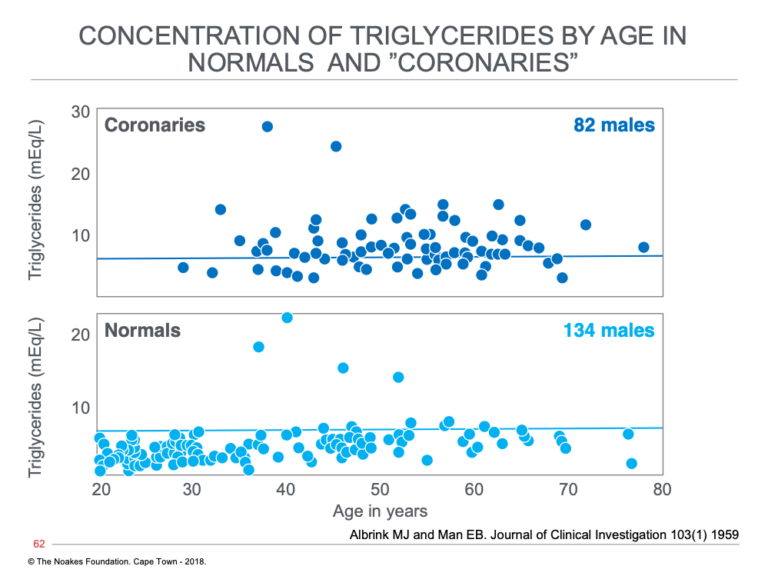
Figure 2: The distribution at different ages of blood triglyceride concentrations in persons without (normals) and those with (coronaries) diagnosed heart attack (acute myocardial infarction). Note that the majority of normal subjects have normal blood triglyceride concentrations, whereas more than 50% of coronaries have elevated blood triglyceride concentrations (above horizontal line at a triglyceride concentration of 8 mEq/L. Reproduced from reference 19.
When Albrink and Man plotted the distribution of these cholesterol and triglyceride concentrations in the two groups, there was much greater overlap in blood cholesterol than in blood triglyceride concentrations (Figure 3), which suggests coronary patients and normal” were more similar in the blood cholesterol than in their blood triglyceride concentrations. From this, Reaven could have drawn only one conclusion: that it cannot be differences in blood cholesterol concentrations driving the greater prevalence of CHD in the coronary patients. He may indeed have wondered: Does the difference in blood triglyceride concentrations explains this difference, and if so, why? This was the research question that would define his legacy.
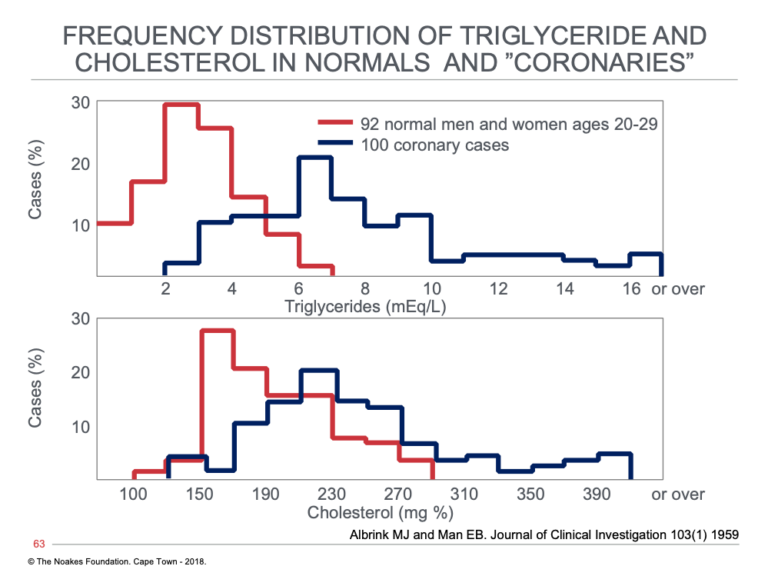
Figure 3: Distribution of blood triglyceride (top) and blood cholesterol (bottom) concentrations in persons without (normals) and those with diagnosed heart attack (acute myocardial infarction) (coronaries). Note that the overlap in these values in much greater for blood cholesterol than for blood triglyceride concentrations. Reproduced from reference 19.
To further analyze the possible association of elevated blood triglyceride concentrations and development of CHD, the authors next compared the percentage of persons with blood triglyceride concentrations higher than 5.5 mEq/L. As shown in Figure 4, the percentage with serum triglyceride concentrations above 5.5 mEq/L rose from about 5% in normal persons between ages 20 and 29 to >70% in those with coronary artery disease and to >85% in 12 subjects who had experienced chest pain during exertion (angina) but who had not suffered a heart attack.
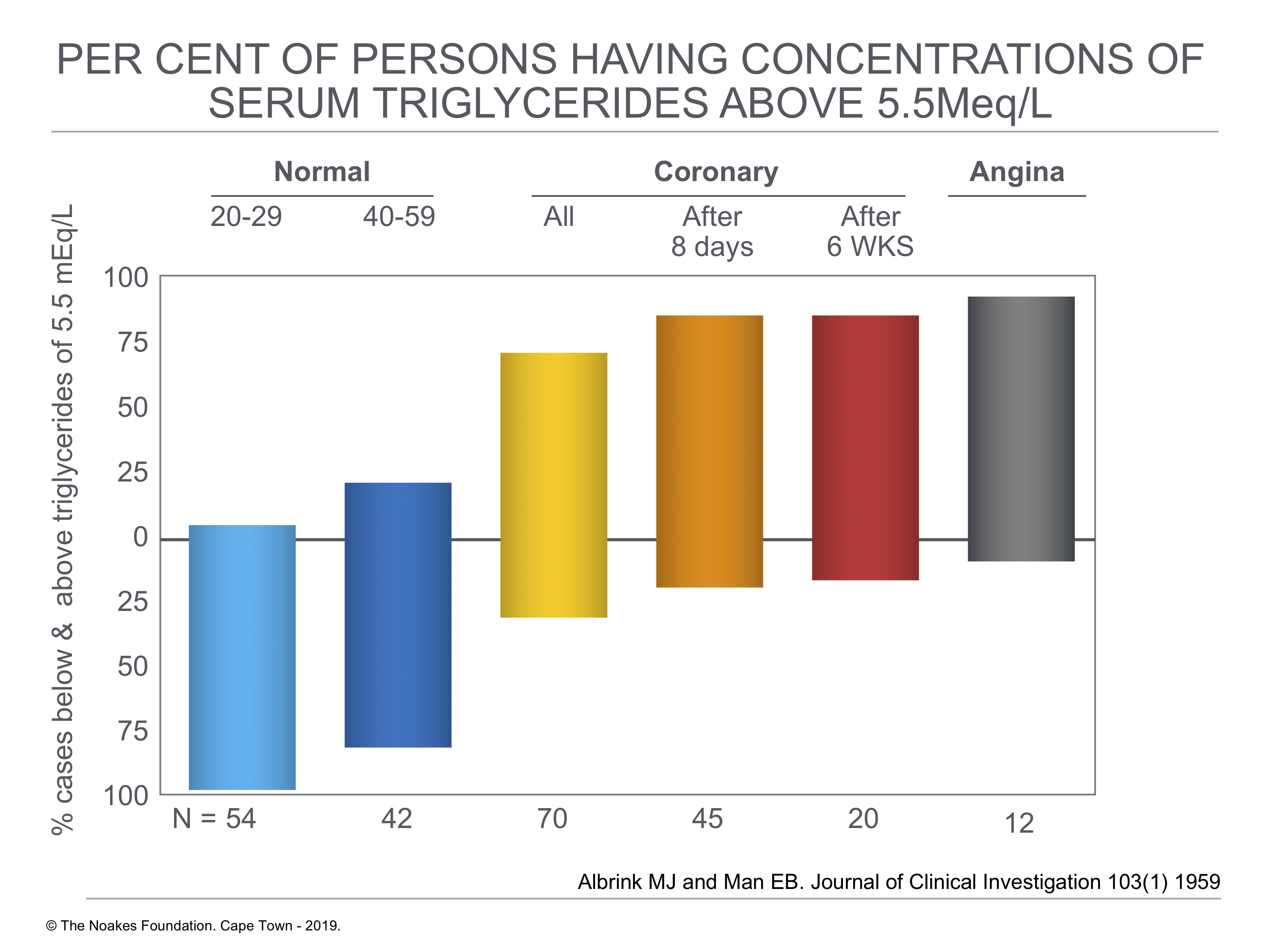
Figure 4: Percentage of cases below and above blood triglyceride concentrations of 5.5 mEq/L in persons without (normal) and those with diagnosed heart attack (acute myocardial infarction) (coronary) and in persons with chest pain during exercise (angina). Note that the percentage with elevated blood triglyceride concentrations rises from about 5% in 20- to 29-years-old normal subjects to above 85% in 12 subjects with angina. Note that the top and bottom extents of the vertical blocks show the % of subjects with (top) and without (bottom) elevated blood triglyceride concentrations. Reproduced from reference 19.
These findings forced the following conclusion:
In summary, elevations of serum triglycerides above 5.5 mEq. per liter (about 160 mg.%) occurred in only 5% of normal young adults, in at most 30% of normal men over 50, and, if the effects of acute illness were excluded, in 85% to 90% of patients with coronary artery disease. Few, if any, other lipid measurements which have been reported effect such a clear-cut separation between the normal and coronary subjects.
And then Albrink and Man added the sentence that probably galvanized Reaven’s attention: “The present report suggests that an error in the metabolism of triglycerides is the lipid abnormality operative in coronary artery disease.”
In their final studies (20, 21) the authors expanded on this theory that blood triglyceride concentrations are more likely to be elevated than are blood cholesterol concentrations in persons with coronary artery disease. Thus, in their 1961 paper (20), they reported, “82 per cent of the patients of all ages with coronary artery disease had high serum triglyceride concentrations.” In contrast, elevated serum cholesterol concentrations “in the absence of elevated triglyceride values characterized only about 10 per cent of the normal population, 10 to 18 percent of the population of the patients with coronary artery disease under fifty years of age, but virtually none of the patients over age fifty” (p. 11).
As a result, they concluded, “The serum triglyceride levels in this series appeared to provide a better separation between normal persons and patients with coronary artery disease than the reported measurements of other serum lipids, and may provide the most accurate single indication yet available of the biochemical defect recognizable in disease of the coronary arteries” (p. 15). They added, “There can be no doubt from the present data that the serum triglyceride concentration is more closely associated with coronary heart disease than is the serum cholesterol concentration” (p. 17).
Only in the very recent past has the key insight of these pioneers — that elevated blood triglyceride concentrations are more effective (associational) predictors of CHD risk than are blood cholesterol markers — begun to be appreciated. The reason for such reluctance is obvious: Whereas the targeted lowering of blood cholesterol concentrations is a highly profitable billion-dollar industry, there are no drugs that effectively lower blood triglyceride concentrations. But there is one simple, cheap and absolutely effective method of which, as I will reveal, these authors were already well aware.
Five years after Albrink and Man’s original publication, Peter Kuo at the Hospital of the University of Pennsylvania published his study of 286 persons with arterial disease and known abnormalities of blood lipid (cholesterol, triglycerides, or both) concentrations (22). He showed that by far the commonest abnormality in 234 (81.8%) of these subjects was what he termed “carbohydrate-induced hyper(tri)glyceridemia.” In fact, he concluded that more than 90% of these patients had the condition:
Although the majority of patients with atherosclerosis in this series were referred to us … (for the investigation) of hypercholesterolemia, only 8.4% were found to have essential familial hypercholesterolemia. More than 90% were found to have carbohydrate-sensitive hyper(tri)glyceridemia with or without hypercholesterolemia. Thus, it is reasonable to assume that with proper dietary preparation and appropriate laboratory studies, a high incidence of carbohydrate-sensitive hyperglyceridemia could be demonstrated in persons with atherosclerosis. (22, p. 92)
Another key finding at about this time was made by Manuel Tzagournis and colleagues (23). They found that the majority of persons with premature CHD before age 49 had elevated blood triglyceride and fasting insulin concentrations, as well as abnormal glucose tolerance. All are markers of impaired insulin sensitivity. They explain, “The high prevalence of abnormal glucose tolerance tests was unexpected because subjects with known clinical diabetes and elevated blood glucose levels were deliberately excluded. This study also disclosed a significant positive correlation between fasting serum triglyceride concentrations and the magnitude of glucose-induced insulin secretion as well as the level of serum cholesterol” (p. 1161).
The study by Tzagournis et al. advanced the finding of Kuo (22) because it suggested a link between carbohydrate-sensitive hypertriglyceridemia, insulin resistance, and premature development of CHD, even in the absence of diagnosed T2DM.
Fast-forward to 1997 and the study by J. Michael Gaziano et al. (24), which found that persons in the highest quartile of blood triglyceride concentrations had a 16-fold higher risk of heart attack than did the group with the lowest blood triglyceride concentrations. The authors could have been repeating the 1959 statement by Albrink and Man when they concluded, “Elevated fasting triglyceride represents a useful marker for risk of coronary heart disease, particularly when HDL levels are considered” (24, p. 2520).
More recently, in 2014, Maria-Agata Miselli and colleagues (25) reported their results from a study in which they followed 1,917 T2DM outpatients over 10 years to evaluate predictors of long-term outcomes. They reported:
In conclusion, we found a direct association between mean triglycerides levels and long-term total mortality risk in older adult type 2 diabetic outpatients; the relationship was significant even after taking into account for the effect of traditional cardiovascular risk factors and pharmacological treatments. This finding suggests that more attention should be given to cardiovascular risk management in type 2 diabetic patients with high triglycerides levels.
The modern literature is now replete with scientific papers reporting precisely the same finding.
It is a pity that the pioneering observations of Albrink and Man have been ignored for so long while an alternate but false hypothesis has been promoted.
Another Important Observation From Albrink and Man
In 1961, Albrink and Man reviewed the data they had collected in the previous 30 years since they began treating T2DM patients in 1931 (20, 21). During this time, they learned that persons with T2DM are at increased risk of developing CHD (26). Importantly, during the course of their study, there had been a significant change in the dietary advice given to all persons with diabetes, including those with T2DM.
Before the discovery of insulin in 1921, it was known that persons with insulin-sensitive T1DM could survive a little longer if they severely restricted their calorie intake and ate a low-carbohydrate diet (27, 28). Thus, in the 1870s, William Morgan wrote, “A diabetic should exclude all saccharine and farinaceous materials from his diet” (29, p. 159). He observed, “Theoretically, the Diabetic should be supplied pretty largely with FAT; and practically it is found that its effect is highly beneficial” (p.162-3).
But the discovery of insulin allowed persons with T1DM to survive when they ate more calories, including more carbohydrate. The result was that, with time, diabetologists began to prescribe diets with progressively higher carbohydrate contents, so that whereas in 1915 the typical diet for a T1DM patient would contain only about 25 grams of carbohydrate per day, already by the 1940 this had increased to 172 grams/day (30, p. 62). Concurrently, diabetologists began to prescribe higher carbohydrate diets also for those with T2DM.
Albrink, Man, and Paul Lavietes proposed that this dietary change had not been without one potentially worrying effect (21). In particular, they argued that this change to a higher carbohydrate diet had produced an increase in average blood triglyceride concentrations in diabetic patients (Figure 5).
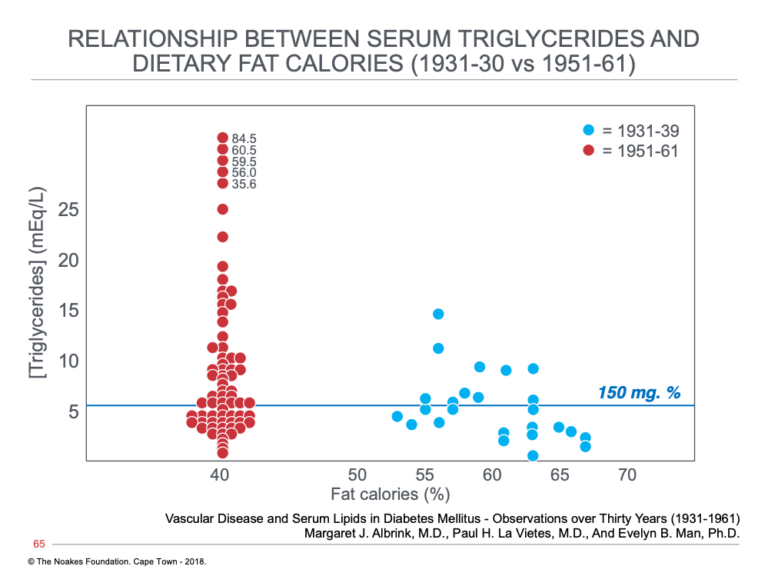
Figure 5: Blood triglyceride concentrations in two populations of patients with T2DM; the one studied from 1931 to 1939, when diets containing between 53 and 67% of total calories as fat were prescribed for persons with T2DM, and the second from 1951 to 1961, when restrictions on dietary carbohydrate intake had been relaxed, causing a reduction in fat intake percentages to about 40%. Note that the number of subjects with elevated blood triglyceride concentrations increased substantially with this increase in carbohydrate intake. Reproduced from reference 21.
Since Albrink and Man were of the opinion that elevated blood triglyceride concentrations rather than higher blood cholesterol concentrations were the better predictors of risk for developing CHD, they were bound to conclude the following:
At least in diabetic persons, the present study suggests that the increased coronary rate over the past 30 years has not been associated with any change in the serum cholesterol, but rather with an increased triglyceride concentration, and the dietary change has been in the direction of lower fat and higher carbohydrate intake rather than the higher fat reported for the country as a whole. Indeed starvation, rigorous caloric restriction, and stringent high fat, low carbohydrate diets were the only form of therapy for diabetes prior to insulin. A similar diet, liberal in fat but low in carbohydrate, was carried over in the early part of the 1930’s when the present study began, but in recent years fat intake has been reduced to that of the country as a whole and carbohydrate intake has been liberalized. Thus the rising triglyceride concentration of diabetics is associated with the trend toward decreasing dietary fat and increasing carbohydrate intake. (21)
As if their first dramatic finding (debunking the lipid hypothesis) was not enough, Albrink and colleagues were now proposing a second theory that would also be completely ignored in the teaching of cardiology and internal medicine, then and now. They proposed that the prescription of a higher carbohydrate diet to persons with T2DM may cause their blood triglyceride concentrations to rise (Figure 5). According to their first controversial theory, this would have to mean that a higher carbohydrate diet would place these persons at increased risk for CHD. Kuo, of course, later provided them with evidence that their theory was correct and a high-carbohydrate diet does indeed elevate blood triglyceride levels.
Kuo noted that others (34, 35) had already observed that very high-carbohydrate diets (80%) could cause blood triglyceride concentrations to increase even in healthy persons (34). But diets containing such large amounts of carbohydrate are unusual, so this finding is not of much practical significance.
Kuo’s next contribution (22, 36) was to establish that humans differ in their sensitivity to this carbohydrate-induced hypertriglyceridemia so that some would develop hypertriglyceridemia with much lower carbohydrate intakes. He wrote:
The salient feature of a patient sensitivity to carbohydrate resides in his ability to develop hyper(tri)glyceridemia on an average American diet, estimated to supply 35% to 40% of total calories largely as refined carbohydrates … (so that) wide fluctuations of the serum triglyceride level in relation to carbohydrate intake is another distinctive feature of carbohydrate-induced hyper(tri)glyceridemia. (22, pp.106-107)
To prove this causal relationship, he placed 64 patients with carbohydrate-sensitive hyper(tri)glyceridemia (CSHT) on a carbohydrate- and sugar-restricted diet for between six and 30 months. The prescribed diet included four elements (22, p. 92):
- Limit carbohydrate intake to 125 to 150 g/day.
- Give complex carbohydrates in place of sugars, fruit juice, and sugar-containing foods. (Sucrose, lactose and fructose had to be totally eliminated from the diet).
- Restrict fats and oils with high short- and medium-chain fatty acid content (milk fat and coconut oil).
- Reduce alcohol consumption.
A typical response to this dietary intervention in one such patient with CSHT is shown in Figure 6.
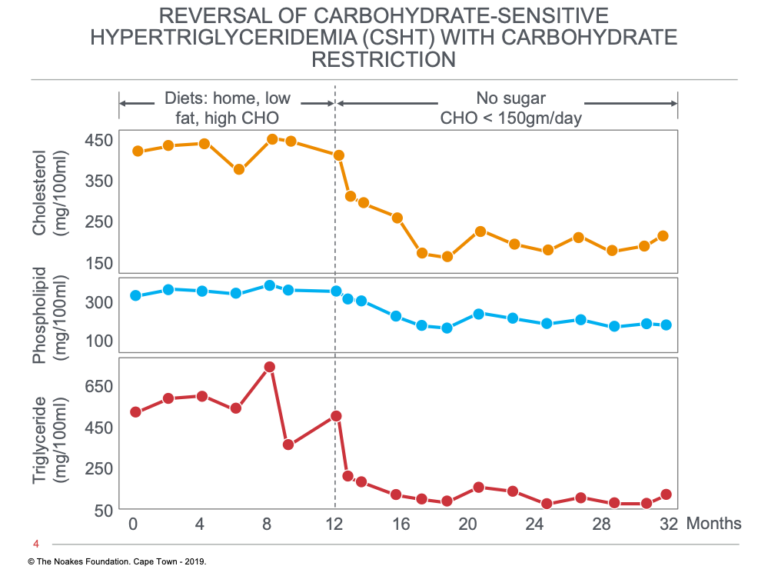
Figure 6: Changes in serum cholesterol, phospholipid, and triglyceride concentrations induced in one subject in response to a dietary change from a low-fat, high-carbohydrate to a low-carbohydrate, high-fat diet with no added sugars at 12 months. Note that all three blood parameters fell on the low-carbohydrate diet and remained low for the remaining 20 months of the study. Reproduced from reference 22.
Figure 7 shows the outcome in all 64 CSHT patients who followed the low-carbohydrate diet for an average of 16.6 months.
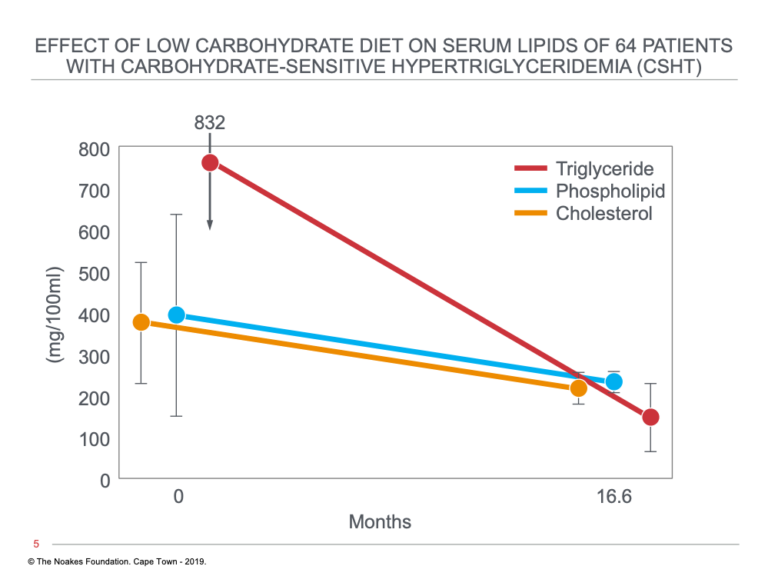
Figure 7: Changes in serum triglyceride, phospholipid, and cholesterol concentrations in 64 subjects with CSHT in response to a dietary change from a low-fat, high-carbohydrate to a low-carbohydrate, high-fat diet with no added sugars at time zero. Note that all three blood parameters fell on the low-carbohydrate diet and remained low for the remaining 16.6 months of the study. Reproduced from reference 22.
An interesting finding shown in Figure 7 is that the low-carbohydrate, no-sugar diet not only lowered blood triglyceride concentrations but also lowered blood cholesterol concentrations.
Another important finding was that sugar and “starch” had quite different effects on blood triglyceride responses in persons with CSHT. Whereas ingesting the same number of calories in the form of sugar substantially raised blood triglyceride concentrations, these concentrations fell steeply when the sugar was replaced with “starch” (36).
What We Knew in the 1960s
So what did we know in 1967 as a result of the research of Albrink, Man, and Kuo, and what have we since forgotten?
- Blood triglyceride concentrations appear to be better predictors of the risk of CHD than blood cholesterol concentrations.
- Blood triglyceride concentrations rise in response to carbohydrate ingestion.
- Individuals differ in the extent to which their blood triglyceride concentrations rise in response to carbohydrate feeding.
- The extent to which blood triglyceride concentrations rise with carbohydrate feeding is one of the best measures of that individual’s degree of insulin-sensitivity/insulin-resistance. Hence, Kuo coined the term “carbohydrate-sensitive hyper(tri)glyceridemia” (CSHT).
- Sugar (sucrose) produced a greater increase in blood triglyceride concentrations than did an equivalent amount of starch (36, 37). This effect was due to the presence of fructose (in sucrose) and resulted from up-regulation of triglyceride synthesis in both liver and adipose tissue (37).
- A carbohydrate-restricted diet lowers blood triglyceride concentrations and may also reduce blood cholesterol concentrations.
Consequences of These Findings
All these findings were most inconvenient, as they were reported at the exact time Ancel Keys and the American Heart Association were beginning to demonize fat, especially saturated fat, as the cause of CHD. Thus, the need arose to glorify carbohydrates, especially “whole” grains, as uniquely healthy.
To achieve this, any finding that carbohydrates may produce undesirable health consequences would have to be suppressed; six decades of the “health-washing” of carbohydrates was about to begin in earnest.
A key component of this carbohydrate “health-washing” would have to be the absolute suppression of any mention that carbohydrate-sensitive hypertriglyceridemia is a — perhaps the key driver of CHD.
This inconvenient fact would need to be hidden over the next 60 years as the false diet-heart and lipid hypotheses became the dominant paradigms directing the teaching and conduct of medical professionals around the globe.
In the following column, we will continue to track Reaven’s journey toward discovering an alternate explanation for CHD.

Professor T.D. Noakes (OMS, MBChB, MD, D.Sc., Ph.D.[hc], FACSM, [hon] FFSEM UK, [hon] FFSEM Ire) studied at the University of Cape Town (UCT), obtaining a MBChB degree and an MD and DSc (Med) in Exercise Science. He is now an Emeritus Professor at UCT, following his retirement from the Research Unit of Exercise Science and Sports Medicine. In 1995, he was a co-founder of the now-prestigious Sports Science Institute of South Africa (SSISA). He has been rated an A1 scientist by the National Research Foundation of SA (NRF) for a second five-year term. In 2008, he received the Order of Mapungubwe, Silver, from the President of South Africa for his “excellent contribution in the field of sports and the science of physical exercise.”
Noakes has published more than 750 scientific books and articles. He has been cited more than 16,000 times in scientific literature and has an H-index of 71. He has won numerous awards over the years and made himself available on many editorial boards. He has authored many books, including Lore of Running (4th Edition), considered to be the “bible” for runners; his autobiography, Challenging Beliefs: Memoirs of a Career; Waterlogged: The Serious Problem of Overhydration in Endurance Sports (in 2012); and The Real Meal Revolution (in 2013).
Following the publication of the best-selling The Real Meal Revolution, he founded The Noakes Foundation, the focus of which is to support high quality research of the low-carbohydrate, high-fat diet, especially for those with insulin resistance.
He is highly acclaimed in his field and, at age 67, still is physically active, taking part in races up to 21 km as well as regular CrossFit training.
References
- Reaven GM. Why Syndrome X? From Harold Himsworth to the insulin resistance syndrome. Cell Metab. 1(2005): 9-14.
- Cran B. The Academic Mob and Its Fatal Toll. Quillette. 2 March 2018.
- Therese S, Martin B. Resist scientist! Countering degradation rituals in science. Prometheus 32(2015): 203-220.
- Noakes TD, Sboros M. Real food on trial: How the diet dictators tried to destroy a top scientist. U.K.: Columbus Publishing Ltd., 2019.
- U.S. Senate Select Committee on Nutrition and Human Needs. Dietary Goals for the United States, 2nd ed. Washington, D.C., U.S.: Government Printing Office, 1977. See more.
- Fox CS, Pencina MJ, Melgs JB, et al. Trends in the incidence of type 2 diabetes mellitus from the 1970s to the 1990s. The Framingham Heart Study. Circulation 113(2006): 2914-2918.
- Mitchell J. Heart and circulatory disease deaths in under 75’s see first sustained rise in 50 years. British Heart Foundation 13 May 2019. Available here.
- Hill JA, Agewell S, Baranchuk A, et al. Medical Misinformation: Vet the message. J Amer Heart Assoc. 18(2009). Available here.
- Reaven GM. Banting lecture 1988. Role of insulin resistance in human disease. Diabetes 37(1988): 1595–1607.
- Himsworth HP. Diabetes mellitus: Its differentiation into insulin sensitive and insulin insensitive types. Lancet 1(1936):127–130.
- Banting FG, Best CH. The internal secretion of the pancreas. J Lab Clin Med. 7(1922): 465–480.
- Hodgkin DC. The Banting Memorial Lecture, 1972. The structure of insulin. Diabetes 21(1972): 1131-1150.
- Himsworth H. The syndrome of diabetes and its causes. Lancet 253(1949): 465-473.
- National Diabetes Data Group. Classification and diagnosis of diabetes mellitus and other categories of glucose intolerance. Diabetes 28(1979): 1039-1057.
- Reaven GM, Lithell H, Landsberg L. Hypertension and associated metabolic abnormalities—the role of insulin resistance and the sympathoadrenal system. N Engl J Med. 334(1996): 374–381.
- Reaven G. Insulin resistance and coronary heart disease in nondiabetic subjects. Arterioscler Thromb Vasc Biol. 32(2012): 1754-1759.
- Yalow RS, Berson SA. Immunoassay and endogenous plasma insulin in man. J Clin Invest. 39(1960): 1157-1175.
- Reaven G, Strom TK, Fox B. Syndrome X. The silent killer. The new heart disease risk. New York, NY: Simon and Schuster, 2001.
- Albrink MJ, Man EB. Serum triglycerides in coronary artery disease. Arch Intern Med. 103(1959): 4-8.
- Albrink MJ, Meigs JW, Man EB. Serum lipids, hypertension and coronary artery disease. Am J Med. 31(1961): 4-23.
- Albrink MJ, Lavietes PH, Man EB. Vascular disease and serum lipids in diabetes mellitus: Observations over thirty years (1931-1961). Ann Intern Med. 58(1963): 305-323.
- Kuo PT. Hyperglyceridemia in coronary artery disease and its management. JAMA 201(1967): 87-94.
- Tzagournis M, Chiles R, Ryan JM, et al. Interrelations of hyperinsulinism and hypertriglyceridemia in young patients with coronary heart disease. Circulation 38(1968): 1156-1163.
- Gaziano JM, Hennekens CH, O’Donnell CJ, et al. Fasting triglycerides, high-density lipoprotein, and risk of myocardial infarction. Circulation 96(1997): 2520-2525.
- Miselli M-A, Nora ED, Passaro N, et al. Plasma triglycerides predict ten-years all-cause mortality in outpatients with type 2 diabetes mellitus: A longitudinal observational study. Cardiov Diabetol. 13(2014): 135.
- Root, HF, Bland EF, Gordon WH, et al. Coronary atherosclerosis in diabetes mellitus. JAMA 113(1939): 27-30.
- Henderson G. Court of last appeal – the early history of the high-fat diet for diabetes. J Diabetes Metab. 7(2016): 8.
- Westman EC, Yancy WS, Humphreys M. Dietary treatment of diabetes mellitus in the pre-insulin era (1914-1922). Perspectives Biol Med. 49(2006): 77-83.
- Morgan W. Diabetes mellitus: Its history, chemistry, anatomy, pathology, physiology and treatment. London: The Homeopathic Publishing Company, 1877.
- Joslin EP. A diabetic manual for the mutual use of doctor and patient. Philadelphia: Lea and Febiger, 1941.
- Allen FM, Stillman E, Fitz R. Total dietary regulation in the treatment of diabetes. Monograph 11. Rockefeller Institute for Medical Research, 1919.
- Rabinowitz IM. Experiences with a high carbohydrate low calorie diet for the treatment of diabetes mellitus. Can Med Assoc J. 23(1930): 489-498.
- Somogyi M. Exacerbation of diabetes by excessive insulin action. Am J Med. 26(1959): 169-191.
- Ahrens EH, Hirsch J, Oette K, et al. Carbohydrate-induced and fat-induced lipemia. Trans Assoc Am Physicians 74(1961): 134-146.
- Hatch FT, Arell LL, Kendall FE. Effects of restriction of dietary fat and cholesterol upon serum lipids and lipoproteins in patients with hypertension. Am J Med. 19(1955): 48-60.
- Kuo PT, Bassett DR. Dietary sugar in the production of hyperglyceridemia. Ann Intern Med. 62(1965): 1199-1212.
- Kuo PT, Feng L, Cohen NN, et al. Dietary carbohydrates in hyperlipemia (hyperglyceridemia); hepatic and adipose tissue lipogenic activities. Am J Clin Nutr. 20(1967): 116-125.