Science Challenging the Somatic Mutation Theory: Nuclear-Cytoplasm Transfer Experiments
The gene theory of cancer is challenged by a growing number of inconsistencies (23-27). Chief among these are the findings from nuclear-cytoplasm transfer experiments. Thomas Seyfried recently reviewed massive evidence showing that nuclei obtained from cancer cells—which presumably contain cancer-driver mutations—cannot drive carcinogenesis when placed in the cytoplasm from normal cells (28). On the other hand, tumors could develop after the placement of a normal nucleus into a tumor cytoplasm. Moreover, replacement of tumor mitochondria with normal mitochondria suppressed growth and metastasis of breast-cancer cells, whereas the opposite was the case when tumor mitochondria replaced normal mitochondria (28).
Figures 1 and 2 illustrate the findings from two of these experiments, while Figure 3 summarizes the findings from a broad range of nuclear-cytoplasm transfer experiments. It is clear that the findings from these experiments are more consistent with the mitochondrial metabolic theory (MMT) than with the somatic mutation theory (SMT), indicating that it is the defects in mitochondria of the cytoplasm rather than defects in the genome of nucleus that underlie the origin of cancer.
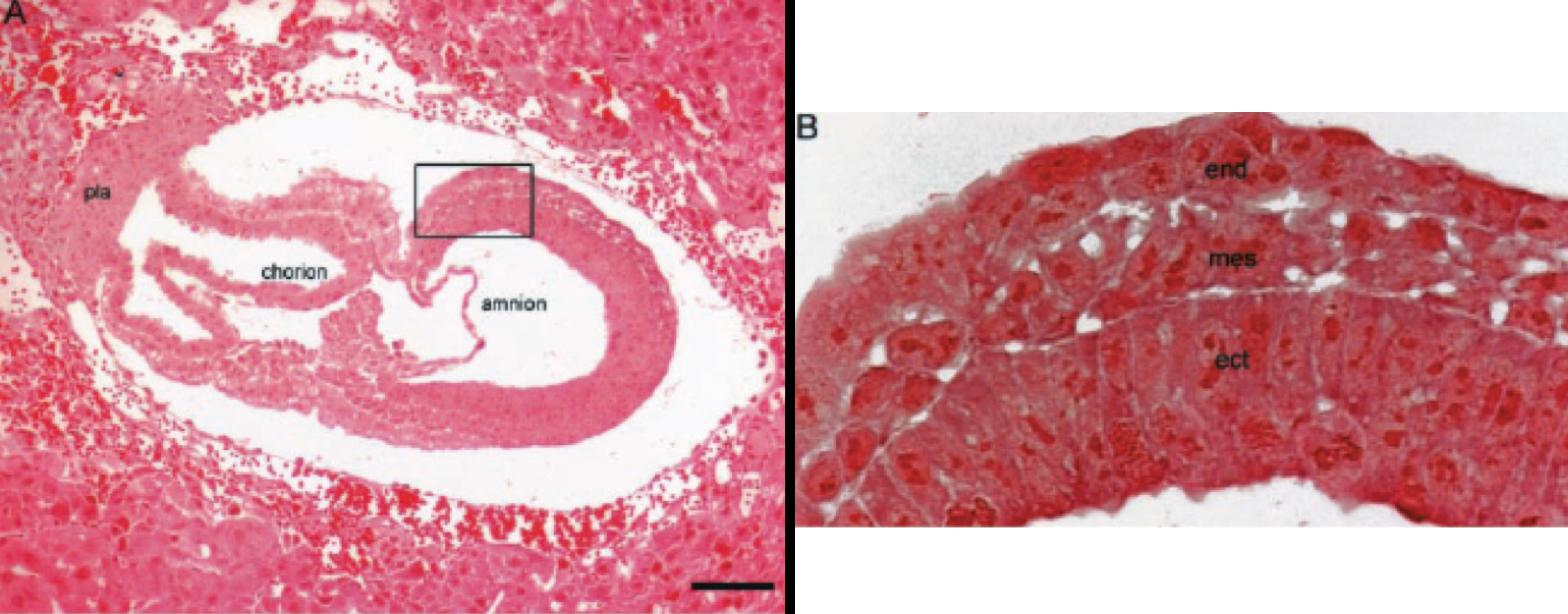
Figure 1. Nuclei from brain tumors support normal mouse embryonic development. Republished with permission from (29).
Figure 1A depicts the H&E staining of a mouse embryo (embryonic day, E-7.5) derived from a cell containing the nucleus from a medulloblastoma tumor. Figure 1B shows the boxed area in A (at a higher magnification), showing the three germ layers; ecto-placental cone (pla); embryonic endoderm (end); embryonic mesoderm (mes); embryonic ectoderm (ect).
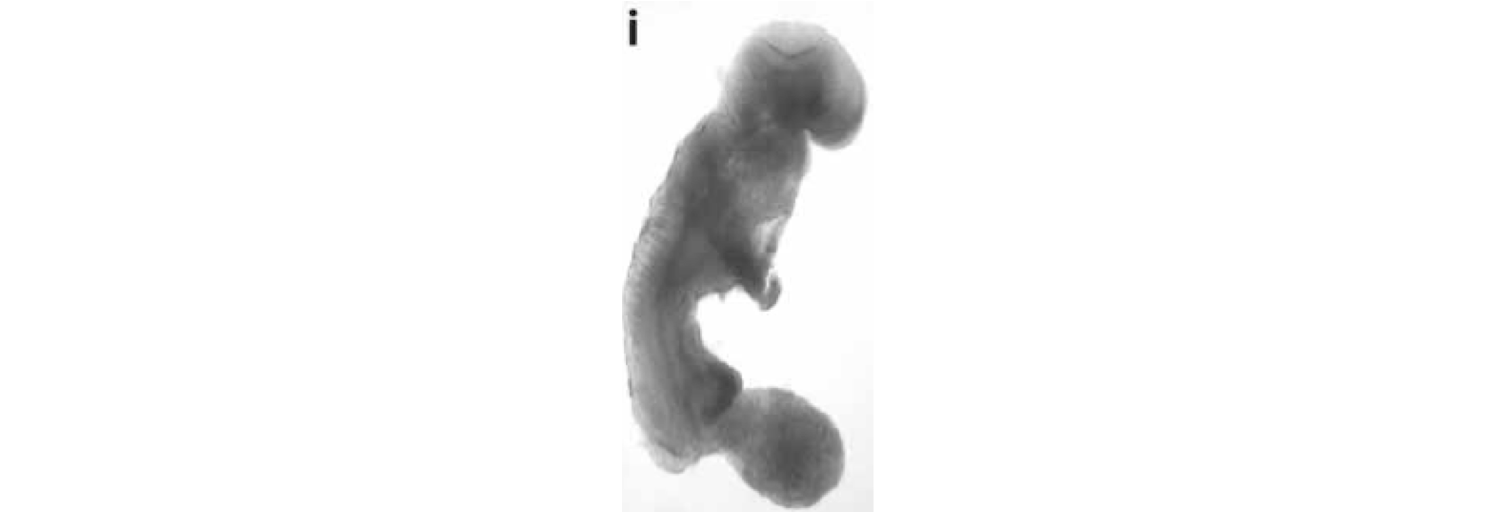
Figure 2. Mouse embryo cloned from tumor cell nucleus. Republished from (31) with permission.
The cytoplasm will contain normal mitochondria. The results show that a nucleus derived from a brain tumor can direct normal embryonic development when implanted into normal cytoplasm. Although the medulloblastoma-derived embryos aborted, none exhibited uncontrolled proliferation resembling tumorigenesis (29). These findings are inconsistent with the SMT.
Figure 2 shows an E-9.5 mouse embryo cloned from a melanoma-derived R545-1 embryonic stem cell. The embryo expressed neural tube closure, a beating heart, and normal limb-bud development consistent with regulated cell growth. The result shows that the nucleus of a malignant melanoma can direct early mouse development when placed into normal cytoplasm containing normal mitochondria. However, irreversible genetic alterations, from the melanoma donor genome, disrupted complete development similar to the situations found in the medulloblastoma experiments (Figure 1), and in Lucke frogs that were cloned from nuclei of renal tumors (30). These findings are also inconsistent with the SMT.
In Figure 3 below, normal cells are shown in green with nuclear and mitochondrial morphology indicative of normal gene expression and respiration, respectively. Tumor cells are shown in red with abnormal nuclear and mitochondrial morphology indicative of genomic instability and abnormal respiration, respectively.
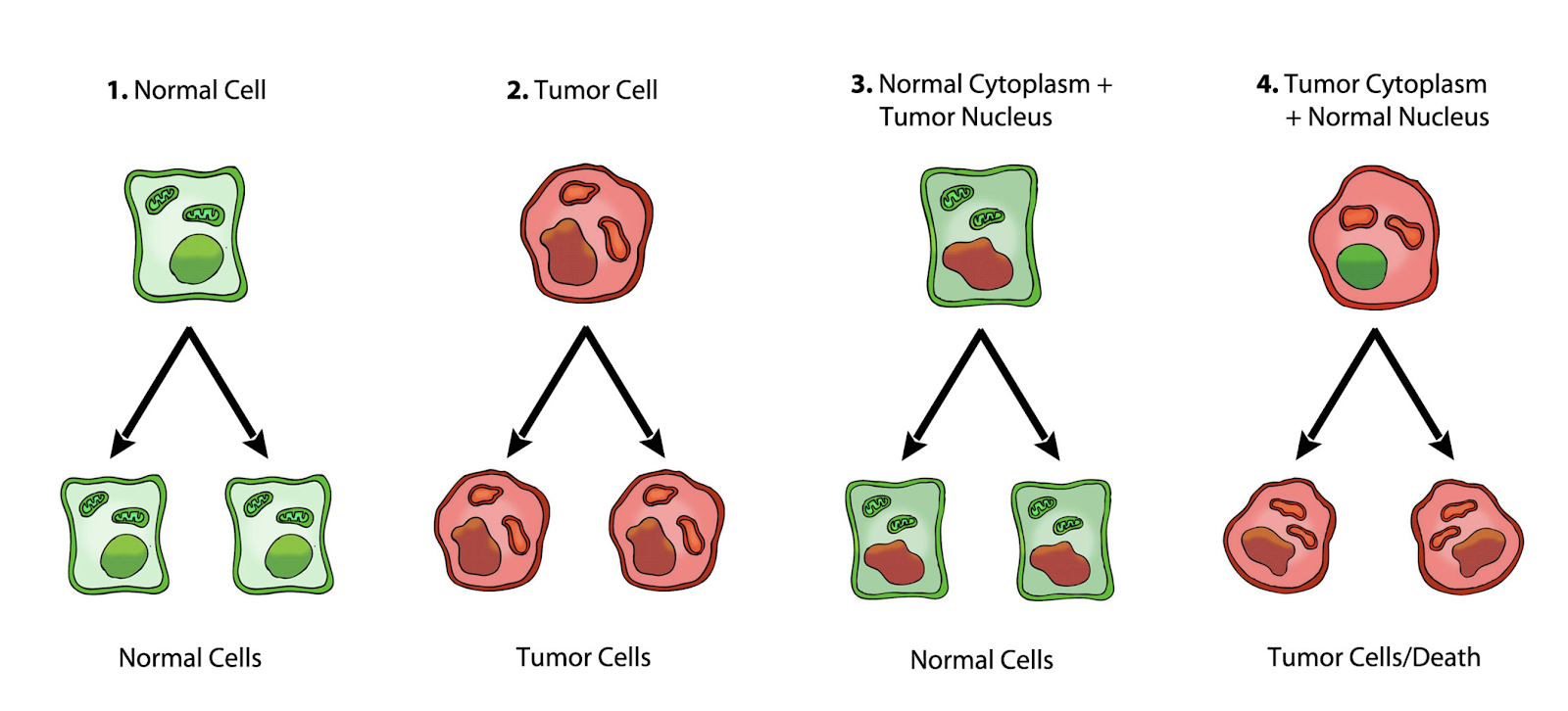
Figure 3. 1) Normal cells beget normal cells (green). 2) Tumor cells beget tumor cells (red). 3) Transfer of a tumor cell nucleus into a normal cytoplasm begets normal cells, despite the presence of the tumor-associated genomic abnormalities. 4) Transfer of a normal cell nucleus into a tumor cell cytoplasm begets dead cells or tumor cells, but not normal cells. Original diagram from Jeffrey Ling and Thomas N. Seyfried, published here with permission.
From Seyfried: “1) Normal cells beget normal cells. 2) Tumor cells beget tumor cells. 3) Transfer of a tumor cell nucleus into a normal cytoplasm begets normal cells, despite the presence of the tumor-associated genomic abnormalities. 4) Transfer of a normal cell nucleus into a tumor cell cytoplasm begets dead cells or tumor cells, but not normal cells. The results suggest that nuclear genomic defects alone cannot account for the origin of tumors, and that normal mitochondria can suppress tumorigenesis.” (28)
Considered collectively, the findings from the nuclear-cytoplasmic transfer experiments provide compelling evidence showing that nuclear somatic mutations alone cannot account for the origin of tumors, and that normal cytoplasm containing mitochondria can suppress tumorigenicity. It is interesting that the findings from the nuclear-cytoplasmic transfer experiments are generally consistent across a broad range of tumor types, animal species, and experimental techniques (28). Importantly, most of the studies were not done to test the somatic mutation theory of cancer but rather to determine the importance of nuclear mutations in directing development. Consequently, data interpretation was largely unbiased.
Several distinguished leaders in the fields of genetics and developmental biology conducted nuclear-cytoplasmic transfer experiments (H. Harris, B. Mintz, R. Sager, J. Morgan, R. Jaenisch) that further supports the validity of the findings. This point is extremely important in light of the recent reproducibility crisis in the field of cancer biology and medicine, where the findings from a broad range of experiments cannot be reproduced (32-34). Although numerous inconsistencies have been documented that undermine the credibility of the SMT (27,35), none of these are as powerful as those presented from the nuclear-cytoplasmic transfer experiments.
Related

Thomas N. Seyfried is professor of biology at Boston College. He received a doctorate in genetics and biochemistry from the University of Illinois—Urbana-Champaign in 1976. He did his undergraduate work at the University of New England, where he recently received the distinguished Alumni Achievement Award. He also holds a master’s degree in genetics from Illinois State University. Seyfried served with distinction in the United States Army’s 1st Cavalry Division during the Vietnam War and received numerous medals and commendations.
He was a postdoctoral fellow in the Department of Neurology at the Yale University School of Medicine and then served on the faculty as an assistant professor in neurology. Seyfried previously served as chair of the Scientific Advisory Committee for the National Tay-Sachs and Allied Diseases Association. He recently received a Lifetime Achievement Award from the Academy of Complementary and Integrative Medicine and the Uncompromising Science Award from the American College of Nutrition for his work on cancer.
He presently serves on several editorial boards, including those for Nutrition & Metabolism, Neurochemical Research, the Journal of Lipid Research, and ASN Neuro. Seyfried has over 180 peer-reviewed publications and is author of the book “Cancer as a Metabolic Disease: On the Origin, Management, and Prevention of Cancer” (Wiley Press).
References
Note: These references include those previously published in “Is Cancer a Genetic or Metabolic Disease? Part 1” and “Part 2.”
- Siegel RL, Miller KD, and Jemal A. Cancer statistics, 2018. CA: A Cancer Journal for Clinicians 68.1(2018): 7-30. Available here.
- Hanahan D and Weinberg RA. Hallmarks of cancer: the next generation. Cell 144.5(2011): 646-674. Available here.
- Vogelstein B, Papadopoulos N, Velculescu VE et al. Cancer genome landscapes. Science 339.6127(2013): 1546-1558. Available here.
- Hou JP and Ma J. DawnRank: discovering personalized driver genes in cancer. Genome Medicine 6.7(2014): 56. Available here.
- Iranzo J, Martincorena I, and Koonin EV. Cancer-mutation network and the number and specificity of driver mutations. Proceedings of the National Academy of Sciences of the United States of America 115.26(2018): E6010-E6019. Available here.
- Fearon ER and Vogelstein B. A genetic model for colorectal tumorigenesis. Cell 61.5(1990): 759-767. Available here.
- Tomasetti C and Vogelstein B. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science 347.6217(2015): 78-81. Available here.
- Vaux DL. In defense of the somatic mutation theory of cancer. BioEssays 33.5(2011): 341-343. Available here.
- McLeod HL. Cancer pharmacogenomics: early promise, but concerted effort needed. Science 339.6127(2013): 1563-1566. Available here.
- Ju J, Zhu A, and Yuan P. Progress in targeted therapy for breast cancer. Chronic Diseases and Translational Medicine 4.3(2018): 164-175. Available here.
- Seyfried TN. Cancer as a Metabolic Disease: On the Origin, Management and Prevention of Cancer. Hoboken, New Jersey: John Wiley & Sons, Inc., 2012. Available here.
- John AP. Dysfunctional mitochondria, not oxygen insufficiency, cause cancer cells to produce inordinate amounts of lactic acid: the impact of this on the treatment of cancer. Medical Hypotheses 57.4(2001): 429-431. Available here.
- Kim A. Mitochondria in cancer energy metabolism: culprits or bystanders? Toxicological Research 31.4(2015): 323-330. Available here.
- Pelicano H, Zhang W, Liu J et al. Mitochondrial dysfunction in some triple-negative breast cancer cell lines: role of mTOR pathway and therapeutic potential. Breast Cancer Research 16.5(2014): 434. Available here.
- Srinivasan S, Guha M, Dong DW et al. Disruption of cytochrome c oxidase function induces the Warburg effect and metabolic reprogramming. Oncogene 35.12(2016): 1585-1595. Available here.
- Stefano GB and Kream RM. Cancer: mitochondrial origins. Medical Science Monitor 21(2015): 3736-3739. Available here.
- Warburg O. On the origin of cancer cells. Science 123.3191(1956): 309-314. Available here.
- Chinopoulos C and Seyfried TN. Mitochondrial substrate-level phosphorylation as energy source for glioblastoma: review and hypothesis. ASN Neuro 10(2018): 1-27. Available here.
- Boveri T. Concerning the origin of malignant tumours by Theodor Boveri. Translated and annotated by Henry Harris. Journal of Cell Science 121. Suppl.1(2008): 1-84. Available here.
- Nowell PC. The clonal evolution of tumor cell populations. Science 194.4260(1976): 23-28. Available here.
- Weinberg RA. The Biology of Cancer. New York City, New York: Garland Science, 2007. Available here.
- Darlington CD. The plasmagene theory of the origin of cancer. British Journal of Cancer 2.2(1948): 118-126. Available here.
- Rous, P. Surmise and fact on the nature of cancer. Nature 183(1959): 1357-1361. Available here.
- Baker SG and Kramer BS. Paradoxes in carcinogenesis: new opportunities for research directions. BMC Cancer 7(2007): 151. Available here.
- Soto AM and Sonnenschein C. The somatic mutation theory of cancer: growing problems with the paradigm? Bioessays 26.10(2004): 1097-1107. Available here.
- Sonnenschein C and Soto AM. Somatic mutation theory of carcinogenesis: why it should be dropped and replaced. Molecular Carcinogenesis 29.4(2000): 205-211. Available here.
- Burgio E and Migliore L. Towards a systemic paradigm in carcinogenesis: linking epigenetics and genetics. Molecular Biology Reports 42.4(2015): 777-790. Available here.
- Seyfried TN. Cancer as a mitochondrial metabolic disease. Frontiers in Cell and Developmental Biology 3(2015): 43. Available here.
- Li L, Connelly MC, Wetmore C et al. Mouse embryos cloned from brain tumors. Cancer Research 63.11(2003): 2733-2736. Available here.
- McKinnell RG, Deggins BA and Labat DD. Transplantation of pluripotential nuclei from triploid frog tumors. Science 165.3891(1969): 394-396. Available here.
- Hochedlinger K, Blelloch R, Brennan C et al. Reprogramming of a melanoma genome by nuclear transplantation. Genes & Development 18.15(2004): 1875-1885. Available here.
- McNutt M. Journals unite for reproducibility. Science 346.6210(2014): 679. Available here.
- Baker M. 1,500 scientists lift the lid on reproducibility. Nature 533.7604(2016): 452-454. Available here.
- Nosek BA and Errington TM. Making sense of replications. eLife 6: e23383, 2017. Available here.
- Sonnenschein C and Soto AM. Theories of carcinogenesis: an emerging perspective. Seminars in Cancer Biology 18.5(2008): 372-377. Available here.
All links accessed Jan. 25, 2019.
Comments on Is Cancer a Genetic or Metabolic Disease? Part 3
Happy to know that this finding is reproductible by others! This makes this study more robust.
Thank you for sharing those. That's incredibly interesting. What happens in the long term when a cell with normal cytoplasm and tumor nucleus duplicates a million times ? We have then a vast majority of "normal" cells with a million "sick" nucleus, however cancer would not be diagnosed, right ?
Is the person living this healthy ? Is it an in-vitro only situation ?
The beauty of these studies was that most were not done to test the somatic mutation theory of cancer but instead were generated to determine the importance of nuclear mutations in directing development. From that perspective, data interpretation was essentially unbiased. But even if you were skeptical, wouldn’t you want to hedge your bet and control everything in your environment that you possibly could, including your body chemistry?
Hi everyone!
I’m so excited! Thanks to Dr. Ahmed for bringing back my Ex-husband and brought great joy to me today! Ahmedutimate@gmail.com is certainly the best spell caster online, if you need your Ex lover back fast! And his result is 100% guarantee….
After 12years of marriage, me and my husband has been into one quarrel or the other until he finally left me and moved to California to be with another woman. I felt my life was over and my kids thought they would never see their father again. I tried to be strong just for the kids but I could not control the pain that torments my heart, my heart was filled with sorrows and pains because I was really in love with my husband. Every day and night I think of him and always wish he would come back to me, I was really upset and I needed help, so I searched for help online and I came across a website that suggested that Dr Ahmed can help get ex back fast. So, I felt I should give him a try. I contacted him and he told me what to do and I did it then he did a (Love spell) for me. 18 hours later, my husband really called me and told me that he miss me and the kids so much, So Amazing!! So that was how he came back that same day, with lots of love and joy, and he apologized for his mistake, and for the pains he caused me and the kids. Then from that day, our Marriage was now stronger than how it was before, all thanks to Dr Ahmed he is so powerful and I decided to share my story on the internet that Dr Ahmed real and powerful spell caster who I will always pray to live long to help his children in the time of trouble, if you are here and you need your Ex back or your husband moved to another woman, do not cry anymore, contact this powerful spell caster now. Here’s his contact:
Email him at: Ahmedutimate@gmail.com
Call / what’s-app him: +2348160153829
Is Cancer a Genetic or Metabolic Disease? Part 3
4