Question
How might impairments in glucose metabolism lead to the development and/or progression of Alzheimer’s disease?
Takeaway
Multiple lines of research suggest defective glucose and/or insulin activity contribute to, and may cause, Alzheimer’s pathology. This same metabolic hypothesis also could explain the significance of known genetic risk factors for Alzheimer’s. Future research will explore whether this metabolic hypothesis translates into effective therapies to prevent and/or treat cognitive impairment.
This 2020 review surveys the literature on the increasingly popular hypothesis that Alzheimer’s disease (AD) is caused, directly or indirectly, by defects in brain metabolism. Interest in the hypothesis has been spurred both by compelling evidence gathered over the past 20 years that metabolic defects in the brain are closely linked to the pathogenesis and symptoms of AD and by the overwhelming failure of the dominant amyloid hypothesis to deliver effective treatments.
It has been known for at least 20 years that diabetics are at least twice as likely as their peers to develop AD. More recent research has illustrated this increased risk is specifically associated with poorly controlled diabetes and particularly with individuals who experience more frequent and severe disease state swings in glucose and insulin levels. This suggests defective patterns of glucose and insulin metabolism may play a role in AD development.
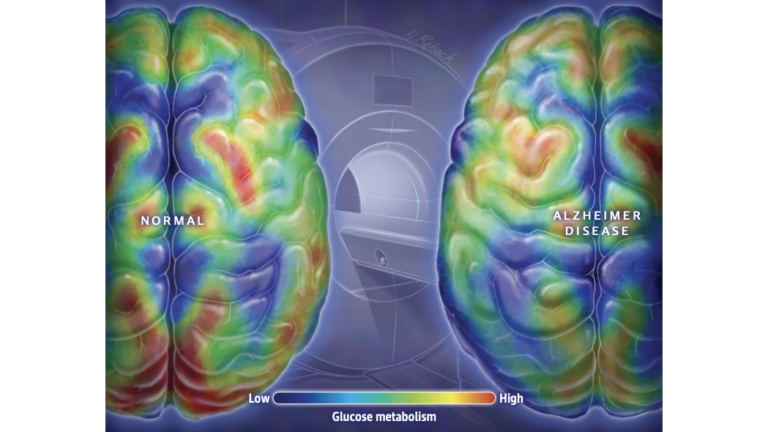
Figure 1: It is well established that the brains of individuals with AD display lower levels of glucose metabolism throughout, and particularly in regions associated with memory and executive function.
Since at least 2003, some researchers have characterized AD as Type 3 diabetes, arguing the brains of individuals with AD show impairments in glucose metabolism and insulin signaling similar to those seen in the periphery (i.e., cells outside the central nervous system) of Type 1 and Type 2 diabetics. Similarly, it is uncontroversial that compared to their peers without cognitive impairment, the brains of AD patients have lower levels of glucose metabolism and insulin signaling, particularly in the regions of the brain associated with memory storage and executive function. Supporters of the Type 3 diabetes hypothesis argue these deficits are not merely biomarkers and instead play a direct causal role in the disease.
As reviewed previously on CrossFit.com, loss of glucose metabolism and/or insulin signaling can directly lead to loss of neuronal function, impaired cell health, and the accumulation of amyloid beta plaques and hyperphosphorylated tau tangles — all established markers of AD progression. Poor metabolic health may also contribute to poor sleep, vascular defects, and inflammation, all of which have been independently linked to AD.
Impairments in glucose metabolism may also explain the significance of the most well-established genetic risk factor for AD. ApoE, which codes for a protein that regulates lipid metabolism, codes for three alleles: ApoE2, ApoE3, and ApoE4. ApoE3 is the most common in the general population. ApoE2 is highly protective against Alzheimer’s, while ApoE4 significantly increases AD risk. Recent mouse research indicates the brains of mice carrying the ApoE2 allele showed increased glucose transport into brain cells and increased glycolytic activity within brain cells. Conversely, ApoE4 carriers have lower levels of glucose transport and glycolysis. As a result, the brains of ApoE2 carriers are more robust amid metabolic challenges, while the brains of ApoE4 carriers more easily become malnourished in response to deficits in glucose availability, glucose transport, and/or insulin signaling. These energy deficiencies have been linked to synapse loss and cognitive impairment.
This hypothesis has not yet yielded an effective treatment for AD. In 2018, a trial testing pioglitazone, which lowers blood glucose levels, was stopped early when it became clear it did not have a significant benefit in patients. Recent and ongoing research is testing intranasal insulin, which delivers insulin directly to the brain and so rapidly ameliorates some forms of insulin resistance; early results have been promising but inconsistent. Other teams have tested the ketogenic diet and other metabolic therapies, which have shown promising but preliminary effects on biomarkers of AD and reversal of AD symptoms. Consequently, the idea that impairments in glucose metabolism can lead to the development of AD remains an interesting and promising hypothesis, but it is yet to be rigorously tested.
Additional Reading
Alzehimer’s disease is Type 3 diabetes: Evidence reviewed
This 2008 review argues the disturbances in glucose and insulin signaling observed in patients with AD can account for the majority of disease morphology and symptomology and are similar to those shown in Type 1 and Type 2 diabetes.
Importance of treatment status in links between Type 2 diabetes and Alzheimer’s disease
This observational study of 1,389 patients from the Alzheimer’s Disease Neuroimaging Initiative found patients with untreated diabetes were more likely to get AD than were nondiabetics or effectively treated diabetics. This same group also displayed the increased levels of amyloid beta and tau that are characteristic of AD.
Sleep disorders in Type 2 diabetes; Aging and pathology cause sleep disruptions and altered metabolism in mouse models of Alzheimer’s disease
The first citation, a 2017 review, summarizes the links between Type 2 diabetes and sleep disorders, noting the frequency and severity of sleep impairments increase with poor glycemic control. The second citation found these metabolically mediated sleep disruptions were associated with AD progression in mice, suggesting such sleep impairments may mediate the relationship between diabetes and AD.
Human ApoE isoforms differentially modulate brain glucose and ketone body metabolism: Implications for early Alzheimer’s disease risk reduction and early intervention; ApoE2-mediated neuroprotective mechanism through regulation of glycolysis
These two studies by the same group found different ApoE alleles (i.e., ApoE2 vs. ApoE3 vs. ApoE4) were associated with different levels of hexokinase expression and glucose transporter activity in the brain. ApoE2 was found to be neuroprotective, supporting sufficient glucose metabolism even when brain cells were metabolically challenged. ApoE4 led to increased vulnerability to metabolic defects. These studies thus suggest the established risk gradations between ApoE alleles may be linked to their impact on brain cell glucose transport and metabolism.
Glucose transporters in brain: In health and Alzheimer’s disease
This 2016 review notes the activity of glucose transporters is decreased in the brains of patients with either Alzheimer’s disease or Huntington’s disease, particularly in the cerebral cortex. These defects in glucose transport lead to deficits in glycolysis and glucose-related ATP production, with brain cells of AD patients producing half the ATP from glucose seen in healthy patients. These defects are associated with an increase in tau pathology, linking defective glucose transport to both AD progression and elements of its chemical pathology.
This mouse study found that accumulation of amyloid in brain cells impairs trafficking of GLUT1, a major glucose transporter, to the cell membrane of brain cells. This suggests amyloid accumulation may itself impair glucose transport, and metabolic defects and amyloid accumulation may form a vicious cycle in the diseased brain.
Evidence for brain glucose dysregulation in Alzheimer’s disease
This 2018 review, drawing from subjects in the Baltimore Longitudinal Study of Aging, found AD patients had characteristically higher circulating levels of blood glucose but lower levels of glucose transporter activity in the brain. The magnitude of these changes correlated to the degree of cognitive impairment, with the first signs of defective glucose metabolism seen several years prior to the emergence of AD symptoms.
Takeda and Zinfadel pharmaceuticals discontinue TOMORROW trial following planned futility analysis
The TOMORROW trial tested the use of pioglitazone, which lowers blood glucose levels, in AD patients. The trial was discontinued prematurely in 2018 when it became clear the drug did not have a significant benefit in patients.
The role of insulin resistance in the pathogenesis of Alzheimer’s disease
In this 2003 review, Suzanne Craft, who would subsequently lead research exploring the use of intranasal insulin as a treatment for AD, argues insulin resistance plays a direct role in AD progression and development, and AD can be linked both to the symptoms of AD and the accumulation of amyloid beta and tau.
Modified ketogenic diet is associated with improved cerebrospinal fluid biomarker profile, cerebral perfusion, and cerebral ketone body uptake in older adults at risk for Alzheimer’s disease: A pilot study; Effect of a ketogenic diet on Alzheimer’s disease biomarkers and symptoms
The first study is a small pilot trial (n=19) that found placing patients on a Mediterranean-ketogenic diet led to improvements in AD biomarkers compared to patients who ate a low-fat diet. The second citation outlines the design for a larger (n=120) randomized controlled trial to test the effects of a Mediterranean-ketogenic diet on both AD symptoms and biomarkers.
Comments on In Alzheimer Research, Glucose Metabolism Moves to Center Stage
And another significant factor may prove to be the modern diet's perturbation of the normal balance of dietary omega-6:omega-3 fats from a ratio of about 1:1 in antiquity to more than 17:1 today. The current heavy reliance on omega-6-rich 'healthy vegetable oils' (which are neither healthy nor vegetable, but rather industrially processed seed oils, rich in linoleic acid) has not only altered the raw materials available to build neuronal cell membranes, but some research suggests the shift may impact/disrupt normal glucose handling by the brain.
https://universityhealthnews.com/daily/memory/aluminum-linked-to-alzheimers-disease/
An additional major contributor to Alzheimer’s is aluminum
In Alzheimer Research, Glucose Metabolism Moves to Center Stage
2