This 2015 review hypothesizes how insulin resistance develops in the brain and cerebral insulin resistance contributes to neurological and metabolic disease.
The brain is known to be an insulin-sensitive organ, with multiple brain regions expressing insulin receptors. Hypothalamus activity, for example, decreases after food intake in what is believed to be an insulin-mediated process; memory formation in the hippocampus is thought to be insulin dependent. These and other known consequences of brain insulin signaling are summarized in Figure 1 below.
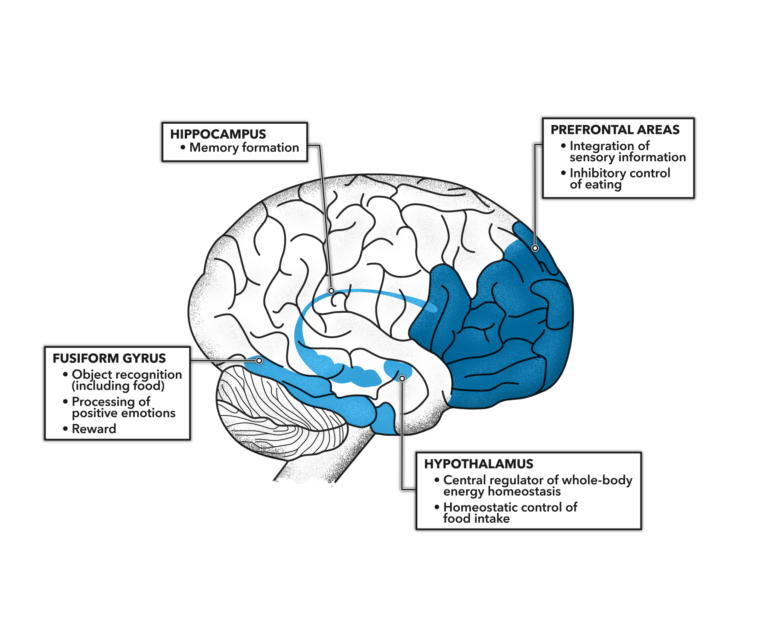
Figure 1: Possible causes of brain insulin resistance. In humans initial evidence exists that brain insulin resistance originates in utero where maternal metabolic derangement interacts with the fetal brain. Genetic background also determines brain insulin sensitivity throughout life. Increasing age and obesity are also linked to impaired transport of insulin across the blood-brain barrier and with insulin resistance in the brain in humans. In particular, enlargement of metabolically unfavorable visceral adipose tissue and raised levels of circulating saturated free fatty acids are associated with brain insulin resistance.
Humans, unlike rats and other “lower” organisms, cannot synthesize insulin directly in the brain. Instead, circulating insulin must be actively transported into the central nervous system (CNS) to reach the brain and other nervous system tissues. In healthy humans, CNS insulin levels are approximately one-tenth of the levels seen in the blood.
Insulin resistance, however, impairs insulin transport into the CNS. Counterintuitively, insulin-resistant individuals, who are often hyperinsulinemic (i.e., have high levels of circulating insulin), can have lower levels of CNS insulin than healthy individuals with lower blood insulin levels. Low CNS insulin levels have also been associated with obesity, diabetes, Alzheimer’s disease, and aging.
These reduced levels of CNS insulin may explain some of the behavioral and metabolic consequences of obesity and the metabolic syndrome. Insulin stimulation of the prefrontal cortex, for example, is responsible for inhibition of food intake (i.e., the urge to stop eating); this response is reduced or eliminated entirely in obese and insulin-resistant individuals. Studies of intranasal insulin, which travels directly to the brain, support this understanding, showing increased CNS insulin levels reduce food intake and can support weight loss when given repeatedly. This may similarly explain the lack of association between serum insulin levels and satiety (i.e., the sensation of fullness) in the obese, and similar degradations of eating behavior and metabolic control that can occur alongside aging.
Preliminary evidence also suggests brain insulin signaling increases whole-body insulin sensitivity. Insulin action in the brain, potentially via effects on the autonomic nervous system, may increase the insulin sensitivity of liver, fat, and muscle cells. This remains a hypothesis, however, as existing research lacks the precision to clearly and specifically characterize these linkages.
In sum, this review argues brain insulin resistance is both a consequence and cause of metabolic distress, and impaired brain insulin signaling can account for both the difficulty of reversing obesity and some of its neurological and metabolic comorbidities.
Impaired Insulin Action in the Human Brain: Causes and Metabolic Consequences